|
|
|
|
|
|
|
|
2.8 Computerberechnungen von lösungsmittelzugänglichen Oberflächen in Proteinen mit bekannter Struktur |
3. Experimenteller Teil
3.1 Reagenzien und Proteine
Die verwendeten Chemikalien, Reagenzien und Lösungsmittel waren generell von p. A. Qualität oder von höchster kommerziell erhältlicher Reinheit:
Acetonitril, Ammoniak (25%), Ammoniumacetat, Ammonium- und Natriumdihydrogenphosphat, Dimethylsulfoxid (DMSO), Eisessig, Ethylendiamintetraessigsäure (EDTA), Guanidin Hydrochlorid, Kupfer(I)chlorid, 2-Mercaptoethanol, Methanol, Natriumdodecylsulfat (SDS), Natriumnitrit, 1-Octanol, Salzsäure, Trifluoressigsäure (UVASOL): Merk
3-Aminopyridin, Arsen(III)chlorid, p-Aminophenylarsonsäure, 6-Aza-2-thiothymin (ATT), Bromcyan (5M Lösung in Acetonitril), 2-Chlor-4,6-diamino-1,3,5-triazin, d6-DMSO, Deuteriumoxid, 2,6-Dihydroxyacetophenon (DHAP), L--Glutamyl-L-cysteinyl-glycin (GSH), Bis-(L--glutamyl)-cystinyl-bisglycin (GSSG), 4-Hydroxy--cyanozimtsäure (HCCA), 4-Vinylpyridin: Aldrich
4-Aminophenylarsonige Säure: Tilman Schmachtel, AG Ullrich, Universität Konstanz
Glycin, Harnstoff, Tris-(hydroxymethyl)aminomethan (Tris): Roth
pH-Eichpuffer 4.0, 7.0, 9.0: Riedel de Häen
Ammoniumhydrogencarbonat, Iodacetamid (IAA): Fluka
Acrylamid, Methylenbisacrylamid: Serva
Dithiothreitol, Coomassie Brilliant Blue-G250, N,N,N',N'-Tetramethylethylendiamin: Sigma
Tris-(2-carboxyethyl)phosphin: Pierce
Entionisiertes Wasser mit dem Widerstandswert von 18.2 M (MilliQ) wurde aus einer Anlage von Millipore bezogen.
Alle verwendete Modellpeptide, -proteine und Enzyme standen als Lyophilisate in Milligramm-Mengen zur Verfügung und wurden bei -18C gelagert:
Oxytocin, (Arg8)-Vasopressin (bovine): Bachem
SP-C(1-14): Rapp Polymere (Tübingen)
SP-C: synthetisch hergestellt [138]
Insulin (bovine), BPTI (Aprotonin), Trypsin (bovine, TPCK behandelt): Sigma
rhM-CSF(4-221A): Chiron Corp., Emeryville, CA.-USA
-Chymotrypsin (bovine): Serva
Endoprotease Glu-C (V8-Protease): Boehringer-Mannheim
3.2 Synthesen von Derivaten der Arsonigen Säure
3.2.1 Pyridin-3-arsonigsäuredichlorid Monochlorhydrat (20) [120]
7.5 g (80 mmol) 3-Aminopyridin werden unter Rühren und Eiskühlung in 30 ml konzentrierter HCl suspendiert. Nacheinander werden 1.2 g Cu(I)Cl (12 mmol) und 30 g (165 mmol) AsCl3 zugegeben. Zu der dickflüssigen, graugrünen Suspension werden unter Rühren und Eiskühlung 8.7 g NaNO2 - gelöst in 12.5 ml H2O - zugetropft. Es setzt sofort starke Gasentwicklung ein (Nitrose Gase und N2) und die Suspension verfärbt sich schwarzgrün. Nach 15 h bei 25 °C wird über eine Umkehrfritte das Reaktionsprodukt von CuCl und NaCl abgetrennt. Das so erhaltene Pyridin-3-arsintetrachlorid (Mutterlauge) wird in einen Kolben überführt und durch Einleiten von SO2 (1 h, 25 °C) zu As(III) reduziert. Der entstandene violette, kristalline Niederschlag wird abgesaugt und sofort aus 300 ml MeOH umkristallisiert. Die so erhaltenen violetten, kristallinen Nadeln werden durch Sublimation (48 h, HV, 120 C Badtemperatur) in einen farblosen, kristallinen Feststoff überführt.
Ausbeute: 3.1 g (17%), Lit: 75%
Schmelzpunkt: 207-210 C, Lit.: 217-230 C
Rf (Kieselgel, MeOH/CHCl3 1:1) = 0.64; Rf(Cellulose, 4% Na-Citrat) = 0.77
1H-NMR: alle ein Proton: 8.08 (t), 8.82 (d), 9.09 (d), 9.20 (s)
EI-MS (70 eV, 25 C) m/z (%): 35 (48), 37 (13), 51 (47), 75 (8), 78 (100), 100 (19), 110 (13), 135 (4), 145 (3), 188 (13), 223 (34, MH+)
FAB-MS (Glycerin/DMSO/Eisessig 1:1:1) m/z (%): 75 (62), 154 (30), 165 (25), 170 (30), 244 (100, Gly.-MH+), 336 (50, 2 Gly.-MH+)
3.2.2 Anhydrid der Pyridin-3-arsonigen Säure (16) (PYR) [120]
weitere Bezeichnungen: 3-Arsenoso-pyridin, Pyridin-3-arsinoxid
Zu einer Lösung von 3 g (10.8 mmol) Pyridin-3-arsonigsäuredichlorid Monochlorhydrat in 7.5 ml H2O (40 C) werden unter Eiskühlung 3.5 ml 25% NH3 (55 mmol) zugetropft. Nach längerem Stehenlassen bei 4 C scheidet sich ein feinkristalliner, farbloser Niederschlag an der Glaswandung ab. Dieser wird abgesaugt, mit wenig Diethylether gewaschen und getrocknet.
Ausbeute: 1.6 g (80%), Lit: 91%
Schmelzpunkt: 119-123 C, Lit.: 118-119 C
Rf (Kieselgel, MeOH/CHCl3 1:1) = 0.85
1H-NMR: 6.97 (s, OH), 7.47 (m, 1H), 8.14 (m, 1H), 8.63 (m, 1H), 8.75 (m, 1H)
UV (MeOH/H2O 9:1): ![]() max = 258 nm,
max = 258 nm, ![]() max = 6100 l mol-1 cm-1
max = 6100 l mol-1 cm-1
EI-MS (70 eV, 170 C) m/z (%): 78 (5), 91 (10), 152 (10), 169 (18, MH+), 231 (41), 260 (16). 309 (4), 321 (10), 429 (39), 507 (57), 598 (1), 676 (1)
FAB-MS (Glycerin/DMSO/Eisessig 1:1:1) m/z (%): 154 (41), 170 (40), 231 (36), 244 (100, Gly.-MH+), 336 (12, 2 Gly.-MH+), 487 (4, 2 Gly.-2 M + H+), 638 (3, 2 Gly.-3 M + H+)
ESI-MS (2% AcOH/MeOH 9:1) m/z (%): 339 (8, M2H+), 354 (16, Me-Ester von M2H+), 508 (62, M3H+), 523 (14, Me-Ester von M3H+), 677 (100, M4H+), 846 (55, M5H+), 1015 (40, M6H+), 1184 (16, M7H+), 1353 (17, M8H+), 1522 (16, M9H+), 1691 (8, M10H+), 1860 (4, M11H+)
Elementaranalyse: C9H9AsN6O 1 H2O (187.03 g/mol)
berechnet C: 32.11% H: 3.20% N: 7.49%
gefunden C: 32.25% H: 2.76% N: 7.55%
3.2.3 p-(4,6-Diamino-1,3,5-triazin-2-yl)aminophenylarsonsäure (25) [124,125]
weitere Bezeichnungen: N-(4,6-Diamino-s-triazin-2-yl) arsanilic acid, p-Melaminylbenzenearsonic acid, p-[(4,6-Diamino-1,3,5-triazin-2-yl)amino]phenylarsonic acid
Synonyme: Friedheim 4289, Melarsen, 2224 R.P. [152]
9.5g (65 mmol) noch feuchtes 2-Chlor-4,6-diamino-1,3,5-triazinwerden in 180 ml Wasser suspendiert. Die Lösung wird auf 60C erwärmt und 0.9 ml konzentrierte HCl, 0.4 ml 1-Octanol und 14.1g (65 mmol) p-Aminophenylarsonsäure zugegeben. Nach 30 min Rückfluß ist der Rückstand vollständig gelöst. Zur leicht orange gefärbten Lösung werden zwei Spatelspitzen Aktivkohle zugegeben, 5 min unter RF gerührt und heiß abfiltriert. Aus der klaren, farblosen Lösung wird durch Zugeben von 20 ml konz. HCl (Eisbad) das Hydrochlorid ausgefällt. Nach dem Absaugen wird der farblose Niederschlag in 230 ml heißem Wasser suspendiert und bei 70C mit NaOH (fest) neutralisiert (pH 6). Nach langsamem Abkühlen und 30-minütigem Rühren der Suspension bei 25 °C wird der sehr feine, amorphe, farblose Niederschlag vorsichtig abgesaugt und zweimal mit je 25 ml Wasser chlorfrei gewaschen (AgNO3).
Ausbeute: 13.4 g (63%); Lit.: 96%
Schmelzpunkt: > 300 C
Rf(Cellulose, 4% Na-Citrat) = 0.7
1H-NMR: (d6-DMSO, Suspension): 4.7 (breit, NH), 6.43 (s, NH2), 7.56 (d, 2H, CH), 8.01 (d, 2H, CH), 9.27 (s, OH)
MALDI-MS (DHB) m/z (%): 327 (100, MH+), 349 (10), 365 (10)
Elementaranalyse C9H11AsN6O3 ½ H2O (335.15 g/mol)
berechnet C: 32.25 % H: 3.61 % N: 25.08 %
gefunden C: 32.37 % H: 3.60 % N: 25.13 %
3.2.4 p-(4,6-Diamino-1,3,5-triazin-2-yl)aminophenylarsonige Säure Monohydrat (14) (MEL) [124,125]
weitere Bezeichnungen: N-(4-Arsenosophenyl)-1,3,5-triazine-2,4,6-triamine, 2-(p-Arsenosoanilino)-4,6-diamino-s-triazine = p-Melaminylphenylarsine oxide
Synonyme: Friedheim 2065, Melarsen Oxyd, Melox, 3177 R.P [152]
6.0 g (18.5 mmol) p-(4,6-Diamino-1,3,5-triazin-2-yl)aminophenylarsonsäure und 0.3 g KI werden in 71 ml konzentrierter HCl suspendiert. 2.2 g SO2 - in 24 ml konzentrierter HCl gelöst - werden unter Rühren bei 25 °C zugetropft. Aus der braunroten Suspension (I2) scheidet sich ein voluminöser, heller Niederschlag ab. Anschließend wird die Suspension 30 min bei 25 °C gerührt und über Nacht bei 4C aufbewahrt, um die Fällung des p-(4,6-Diamino-1,3,5-triazin-2-yl)aminophenylarsonigsäuredichlorids, das wahrscheinlich als Hydrochlorid vorliegt, zu vervollständigen.
Das Dichlorid wird durch Eingießen der Suspension auf 170 g Eis und durch Zugeben einer Lösung von 62.5 g NaHCO3 in 170 ml Wasser hydrolysiert. Der pH-Werts wird auf 6-7 mit Na2CO3 eingestellt. Die (freie) Arsonige Säure wird abgesaugt, und durch Resuspension des graufarbenen, feinkristallinen Niederschlags in 85 ml Wasser und anschließendes Absaugen gewaschen. Dieser Niederschlag wird in 5-10 ml 2N NaOH unter RF gelöst. Die gelbe, klare Lösung wird mit zwei Spatelspitzen Aktivkohle versetzt, 5 min unter RF gerührt und heiß abfiltriert. Mit 2N Essigsäure wird die Lösung neutralisiert. Aus dem weißen amorphen Niederschlag bilden sich nach 5-7 Tagen bei 4C farblose, feinkristalline Nadeln. Diese werden abgesaugt, zweimal mit 5-10 ml kaltem Wasser gewaschen und getrocknet 0.8 g (20%).
Der aus der Mutterlauge nach 2 Tagen Stehenlassen ausfallende Niederschlag wird abgesaugt, zweimal mit 10 ml kaltem Wasser gewaschen und in 250 ml Wasser umkristallisiert (1.3 g, 33%).
Ausbeute: 2.6 g (53%), Lit.: 78%
Schmelzpunkt: > 300 C
Rf (Cellulose, 4% Na-Citrat): 0.10
1H-NMR (DMSO-d6): 6.34 (s, NH2), 6.60 (s, NH), 7.46 (d,2,CH) ,7.86 (d,2,CH), 8.98 (d, OH)
UV (MeOH/H2O 9:1): ![]() max = 278 nm,
max = 278 nm, ![]() max = 38400 l mol-1 cm-1
max = 38400 l mol-1 cm-1
EI-MS (70 eV, 270 C) m/z (%): 63 (100), 78 (70), 94 (5), 201 (1.8), 202 (2.5)
FAB-MS (DMSO/Eisessig/Glycerin 1:1:1) m/z (%): 75 (33), 165 (8), 203 (53), 293 (17, MH+), 367 (90, Gly.- MH+), 459 (15, 2 Gly.- MH+), 733 (0.9, 2 Gly.-2 M+H+), 1099 (0.04, 2 Gly.-3 M+H+), 1465 (0.01, 4 Gly.-4 M+H+)
ESI-MS (MeOH/H2O 9:1) m/z (%): 293 (90), 311 (100 MH+), 324 (50, Me-Ester), 476 (25, M2H+ Fragment), 877 (20, M3H+)
MALDI-TOF-MS (HCCA) m/z (%): 211 (30), 293 (60), 311 (85, MH+), 476 (55, M2H+ Fragment), 482 (100, HCCA-MH+), 585 (5, M2H+), 671 (10, 2 HCCA-MH+), 877 (3, M3H+)
Elementaranalyse: C9H9AsN6O 2 H2O (328.13 g/mol)
berechnet C: 32.94% H: 3.99% N: 25.61%
gefunden C: 33.05% H: 4.05% N: 25.54%
nach 12 h 120 C: C9H9AsN6O 1 H2O (310.13 g/mol)
berechnet C: 34.90% H: 3.58% N: 27.10%
gefunden C: 34.62% H: 3.56% N: 27.17%
3.2.5 p-Aminophenylarsonige Säure (17) (A-PAA)
weitere Bezeichnungen: p-Aminophenylarsenoxid
Synthese und Charakterisierung (1H-NMR, EI-MS) ist in [119] beschrieben.
UV (MeOH/H2O 9:1): ![]() max = 264 nm,
max = 264 nm, ![]() max = 15600 l mol-1 cm-1
max = 15600 l mol-1 cm-1
ESI-MS (2% AcOH/MeOH 9:1) m/z (%): 184 (100), 201 (20), 258 (60), 351 (15), 368 (5), 624 (20), 808 (15), 1006 (10), 1264 (5), 1447 (4)
MALDI-TOF-MS (HCCA oder ACH) m/z (%): 184 (80), 258 (100), 351 (20), 368 (18), 443 (18), 625 (5), 701 (4) 3.3
3.3 UV-spektroskopische Untersuchungen
Alle photometrischen Messungen wurden mit dem Einstrahl UV/VIS-Photometer UV1202 (Shimadzu) durchgeführt. Es wurden die mitgelieferten Quarzglasküvetten mit 0.4 mm Durchsichtsfenster und 10 mm optischer Weglänge (max. 1500 µl Flüssigkeitsvolumen) verwendet. Die Steuerung und Datenaufnahme erfolgte mit einen 286er-Personal Computer. Die Software UVMONO zur Datenaufnahme wurde von Markus Kalkum zur Verfügung gestellt [117].
3.3.1 Extinktionskoeffizient und Löslichkeit der synthetisierten Arsonigen Säuren
- Zur Bestimmung der langwelligsten Absorption und des Extinktionskoeffizienten in Methanol und Methanol/Wasser-Gemischen wurden von den einzelnen Derivaten MEL, PYR und A-PAA 1 mM Lösungen hergestellt. Für PYR und A-PAA wurden die Lösungen mit dem entsprechenden Lösungsmittel auf c = 10/15/20 µM, für MEL auf c = 10/20/30 µM verdünnt und UV-Spektren im Wellenlängenbereich von = 201-339 in Schritten von 2 nm aufgezeichnet. Die so ermittelte langwelligste Absorption
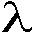 max und der nach dem Bougour-Lambert-Beerschen Gesetz (E( ) =
max und der nach dem Bougour-Lambert-Beerschen Gesetz (E( ) = 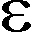 () c d) ermittelte Extinktionskoeffizient max wurde gemittelt. Zur Bestimmung der maximal löslichen Konzentration wurden Lösungen/Suspensionen mit einer Sollkonzentration von 0.5 M an MEL, PYR und A-PAA hergestellt und 4 h bei 25 C inkubiert. Von den Überständen dieser Lösungen wurde dann UV-spektroskopisch die Konzentration bestimmt.
() c d) ermittelte Extinktionskoeffizient max wurde gemittelt. Zur Bestimmung der maximal löslichen Konzentration wurden Lösungen/Suspensionen mit einer Sollkonzentration von 0.5 M an MEL, PYR und A-PAA hergestellt und 4 h bei 25 C inkubiert. Von den Überständen dieser Lösungen wurde dann UV-spektroskopisch die Konzentration bestimmt. - Zur Bestimmung der langwelligsten Absorption und des Extinktionskoeffizienten von MEL in rein wäßrigen Systemen wurden für pH 8, pH 6 0.2 mM und für pH 4, pH 2 0.4 mM MEL-Lösungen für die inTab. 14 aufgelisteten Puffersysteme hergestellt. Die Lösungen wurden auf 45 µM MEL mit den entsprechenden Puffersystemen verdünnt und Spektren analog a) aufgezeichnet. In allen Fällen wurde mindestens eine Doppelbestimmung durchgeführt.
Die ermittelten Absorptionsmaxima ![]() max und Extinktionskoeffizienten
max und Extinktionskoeffizienten ![]() max sind in Tab. 14 zusammengefaßt.
max sind in Tab. 14 zusammengefaßt.
Tab. 14: Absorptionsmaximum ( ![]() max) und Extinktionskoeffizient (
max) und Extinktionskoeffizient (![]() max) von MEL, PYR und A-PAA in verschiedenen organischen Lösungsmittel(gemischen) und wäßrigen Puffersystemen.
max) von MEL, PYR und A-PAA in verschiedenen organischen Lösungsmittel(gemischen) und wäßrigen Puffersystemen.
|
Lösungsmittel / Puffer |
MEL |
PYR |
A-PAA |
|||
|
|
|
|
|
|
|
|
|
MeOH |
278 |
39300 |
258 |
(> 3800) |
264 |
12000 |
|
MeOH / H2O 9:1 |
278 |
38400 |
258 |
6100 |
264 |
15600 |
|
10 mM NH4HCO3 pH 8.2 |
273 |
28400 |
n.b. |
n.b. |
n.b. |
n.b. |
|
10 mM NH4OAc pH 6.0 |
273 |
28000 |
n.b. |
n.b. |
n.b. |
n.b. |
|
10 mM NH4OAc pH 4.0 |
275 |
25700 |
n.b. |
n.b. |
n.b. |
n.b. |
|
10 mM Glycin pH 2.0 |
273 |
23800 |
n.b. |
n.b. |
n.b. |
n.b. |
|
50 mM NaH2PO4 pH 8.5(a) |
273 |
31500 |
n.b. |
n.b. |
n.b. |
n.b. |
|
50 mM NaH2PO4 pH 7.0 (a) |
275 |
30100 |
n.b. |
n.b. |
n.b. |
n.b. |
|
50 mM NaH2PO4 pH 6.0 (a) |
275 |
28200 |
n.b. |
n.b. |
n.b. |
n.b. |
|
50 mM NaH2PO4 pH 4.0 (a) |
275 |
22000 |
n.b. |
n.b. |
n.b.. |
n.b. |
|
50 mM NaH2PO4 pH 2.0 (a) |
275 |
21000 |
n.b. |
n.b. |
n.b. |
n.b. |
3.3.2 Stöchiometrie und zeitlicher Verlauf der Modifizierung von Bis-Thiolen mit Melarsen Oxid (MEL)
Folgende Stammlösungen von MEL wurden hergestellt und bei 4 C aufbewahrt: 0.2 mM MEL in (a) 10 mM NH4HCO3 pH 8.2 und (b) in 10 mM NH4OAc pH 6.0; 0.4 mM MEL in (c) 10 mM NH4OAc pH 4.0 und (d) in 10 mM Glycin pH 2.0; (e) 0.2 mM MEL in 1 mM EDTA, 50 mM NaH2PO4 pH 8.5 / 7.0 / 6.0 / 4.0 und 2.0.
Folgende Dithiollösungen wurden verwendet, langfristig bei -20 C und unmittelbar vor der Verwendung bei 4C aufbewahrt: 1 mM DTT und 1 mM DMP in den entsprechenden Puffern (a) bis (d), 1 mM Oxytocin (OT), reduziert mit 5 mM TCEP in 10 mM NH4HCO3 (siehe 3.4.1), 5 mM GSH in 1 mM EDTA, 0.72 M Harnstoff, 50 mM NaH2PO4 pH 8.5.
Die Thiolkonzentration von DMP und DTT wurde vor der Umsetzung mit MEL mit Ellmans Reagenz (3.4.5) bestimmt, um Fehler aufgrund der Autooxidation der Dithiole zu vermeiden. TCEP reagiert mit DTNB, so daß eine analoge Bestimmung für Oxytocin nicht möglich war.
Für alle untersuchten pH-Bereiche und Puffersysteme (a) bis (d) wurden mit DMP, DTT und OT folgende Versuchsreihen durchgeführt:
- Aufnahme des UV-Spektrums (201-339, = 2) von 45 µM MEL (Standard M). Dazu wurden 225 µl der 0.2 mM MEL-Lösungen (pH 8.2 und 6.0) bzw. 113 µl der 0.4 mM MEL-Lösungen (pH 4.0 und 2.0) mit den entsprechenden Puffern in der UV-Küvette auf 1 ml Gesamtvolumen aufgefüllt.
- Aufnahme der UV-Spektren (201-339, = 2) für DMP, DTT und Oxytocin (eingesetzte Konzentrationen 15, 30, 45, 60 und 100 µM; Standard Tn bzw. Standard OTn). Dazu wurden 15, 30, 45, 60 und 100 µl der 1 mM Dithiollösungen mit den entsprechenden Puffern in der UV-Küvette auf 1 ml Gesamtvolumen aufgefüllt.
- Für jedes zu untersuchende molare Verhältnis von MEL zu Dithiol wurde MEL in der UV-Küvette vorgelegt und entsprechend mit dem jeweiligen Puffer verdünnt, so daß nach Zugabe des Dithiols ein Gesamtvolumen von 1 ml und damit eine MEL-Konzentration von 45 µM resultierte. Vor der Zugabe der Dithiolkomponente wurde die Aufzeichnung der zeitliche Änderung der Absorptionswerte für die Wellenlängen des Minimums und Maximums des Differenzspektrums (siehe Tab. 15) gestartet. Für die Umsetzung von MEL mit OT wurden die in Tab. 15 angegebenen Wellenlängen verwendet, ansonsten wurde - der Einfachheit wegen - die zeitliche Änderung der Absorptionswerte bei 267 nm und 299 nm aufgezeichnet.
- Nach ca. 60 s wurde das UV-Photometer geöffnet und das entsprechenden Volumen (siehe Punkt 2) an Dithiol zur MEL-Lösung pipettiert und mit einem Mikrospatel vermischt. Dieser Vorgang dauerte durchschnittlich 15-30 s.
- Nach Erreichen konstanter Absorptionswerte wurde von der Reaktionslösung das UV-Spektrums (201-339, = 2) aufgenommen.
Die Umsetzungen mit GSH erfolgten analog für folgende Konzentrationen: 25, 50, 75, 150, 300, 600 und 1200 µM, jedoch nur im Puffersystem 1 mM EDTA, 0.72 M Harnstoff, 50 mM NaH2PO4 pH 8.5.
Tab. 15: Wellenlängen der Minima und Maxima der Differenzspektren für die quantitative Umsetzung von MEL mit DMP, DTT und (reduziertem) Oxytocin in Abhängigkeit des verwendeten Dithiols und pH-Werts.
|
pH-Wert |
DMP |
DTT |
Oxytocin (red.) |
|||
|
|
|
|
|
|
|
|
|
2.0 |
266.5 |
303 |
265.5 |
303 |
264 |
303 |
|
4.0 |
269 |
302 |
268 |
304 |
267 |
303 |
|
6.0 |
n.b. |
n.b. |
267 |
299.5 |
268 |
301 |
|
8.2 |
267 |
299 |
n.b. (a) |
n.b. (a) |
268 |
301 |
In Abb. 88 sind die Ergebnisse zur chemischen Selektivität und Stöchiometrie (vgl. 2.2.2) für die Reaktion von MEL mit DMP und DTT für verschiedene pH-Werte dargestellt. Experimentell schwierig ist die Bestimmung der ![]() OD299-267 Werte für MEL und DTT, da das DTT-MEL-Addukt bei pH 8 und 6 ausfällt und damit
OD299-267 Werte für MEL und DTT, da das DTT-MEL-Addukt bei pH 8 und 6 ausfällt und damit ![]() OD299-267 unspezifisch zunimmt (Abb. 88 B). In Abb. 89 sind die Ergebnisse für den zeitlichen Verlauf (vgl. 2.2.3) der Reaktion von MEL mit DMP für verschiedene pH-Werte dargestellt.
OD299-267 unspezifisch zunimmt (Abb. 88 B). In Abb. 89 sind die Ergebnisse für den zeitlichen Verlauf (vgl. 2.2.3) der Reaktion von MEL mit DMP für verschiedene pH-Werte dargestellt.
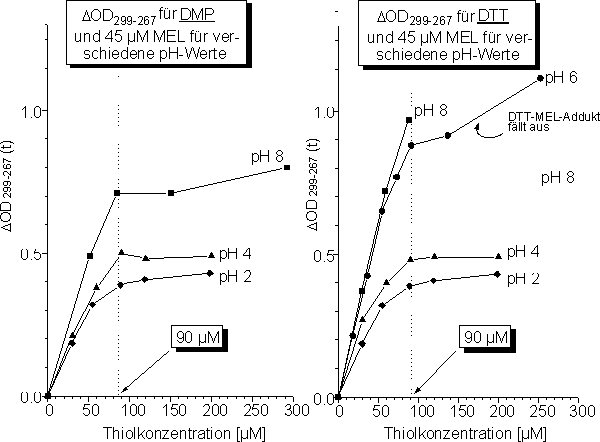
Abb. 88: Umsetzungsgrad von 45 µM MEL mit (A) DMP und (B) DTT in Abhängigkeit der Thiolkonzentration für verschiedene pH-Werte.
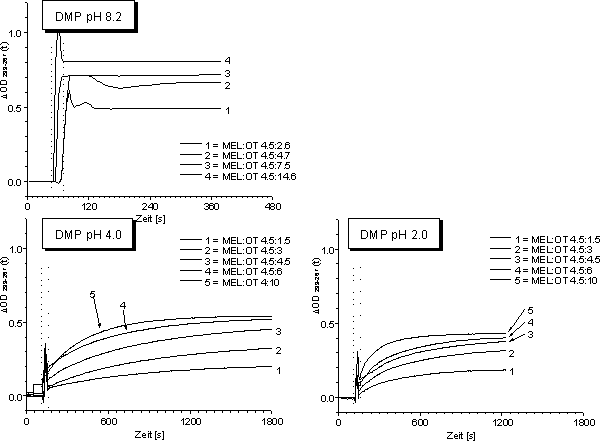
Abb. 89: Zeitlicher Verlauf der Reaktion von 45 µM MEL mit verschiedenen Konzentration an DMP für verschiedene pH-Werte. Die Legende gibt die molaren Verhältnisse wieder. Die gestrichelten vertikalen Linien entsprechen der Zugabe des Dithiols zur MEL-Lösung (Öffnen des UV-Photometers).
3.4 Peptid- und Proteinchemische Umsetzungen
3.4.1 Reduktion von Disulfidbrücken mit 1,4-Dithiothreitol und Tris-(2-carboxyethyl)phosphin
Die Reduktion von Disulfidbrücken erfolgte entweder mit 2-Mercaptoethanol (ME), 1,4-Dithiothreitol (DTT) oder Tris-(2-carboxyethyl)phosphin (TCEP). Alle Reduktionsmittel wurden in Wasser gelöst (250 mM) und die Stammlösungen bei -20 C aufbewahrt. Für die Reduktionen wurden die Peptide/Proteine in folgenden Puffern vorgegeben.
|
Für die durchgeführten Reduktionsreaktionen wurden die lyophilisiert vorliegenden Peptide (Oxytocin, Vasopressin) und Proteine (BPTI, Insulin) auf einer Mikrowaage abgewogen, in den entsprechenden Puffern A-E (siehe Tab. 16) gelöst und mit den jeweiligen Volumina der Stammlösungen der Reduktionsmittel versetzt, so daß die in Tab. 16 angegebenen molaren Verhältnisse (Peptid/Protein zu Reduktionsmittel) und Proteinkonzentrationen (cprotein) bzw. Reduktionsmittelkonzentrationen (credm.) resultierten. Für das verwendete rhM-CSF wurde die nach der Ultrafiltration (siehe 3.6.2) vorliegende Proteinkonzentration bestimmt (siehe 3.6.5) und zu diesen Lösungen die entsprechenden Mengen an Reduktionsmittel zugegeben. Die Reduktion erfolgte unter den in Tab. 16 angegebenen Bedingungen. Die Reaktionslösungen wurden nach erfolgter Reduktion bei -20 C aufbewahrt.
Anmerkungen:
- Die vollständige Reduktion und Denaturierung von rhM-CSF für die Faltungsreaktionen sind in Abschnitt 3.8 beschrieben.
- Die eingesetzten molaren Verhältnisse für die Reduktionen von BPTI, Insulin und rhM-CSF entsprechen einem 20-fachen molaren Überschuß an Reduktionsmittel pro Disulfidbrücke.
- Zur vollständigen Reduktion von BPTI wurden folgende Reduktionsbedingungen untersucht: BPTI (1 µg/µl) wurde in NH4HCO3-Puffer (10 mM, pH 8.2) mit und ohne 6 M Harnstoff gelöst. Zum Teil wurden die Lösungen vor oder nach Zugabe von TCEP (molares Verhältnis BPTI / TCEP 1:60, ctcep = 18 mM) 5 Minuten bei 95 C hitzedenaturiert. Alle Reduktionsansätze wurden 2 h bei 37 C inkubiert und Aliquote davon mit Iodacetamid alkyliert (siehe 3.4.2). Die MALDI-massenspektrometrische Analyse zeigte vollständig reduziertes BPTI sowohl für die Reduktion in 6 M Harnstoff ohne Hitzedenaturierung als auch ohne Harnstoff, aber Hitzedenaturierung in Gegenwart des Reduktionsmittels. Die Hitzedenaturierung in Gegenwart von Harnstoff führt ebenfalls zu vollständig reduziertem BPTI. Jedoch erhält man zusätzlich Carbamoylierungen, die als Signalserien mit einer Massendifferenz von m = 44 in den MALDI-Massenspektren auftreten [153]. In Harnstofflösungen können sich geringe Mengen an Cyanationen bilden [154], die Proteine an den -Aminogruppen der Lysinreste carbamoylieren können [155]. Für die Reduktion bei Raumtemperatur in Gegenwart von 6 M Harnstoff wurden keine Carbamoylierungen beobachtet. Guanidinium Hydrochlorid zeigt keine Zersetzung im Basischen und ist deshalb für die Reduktion unter denaturierenden Bedingungen besser geeignet.
Tab. 16: Übersicht der Reduktionsbedingungen. Für die eingesetzten Peptide/Proteine sind die verwendeten Reduktionsmittel, Puffer und molaren Verhältnisse von Peptid/Protein zu Reduktionsmittel, sowie der entsprechende Abschnitte im Text angegeben. Die Proteinkonzentrationen cprotein und Reduktionsmittelkonzentrationen credm. geben die jeweiligen Endkonzentrationen während der Reduktion an.
|
Peptid/Protein |
Abschn. |
Reduktionsmittel |
Puffer |
mol. Verh. Prot./Redm. |
cprotein[ug/µl] |
credm. [mM] |
Bedingungen |
|||||||
|
für die UV-spektroskopischen Untersuchungen: |
||||||||||||||
|
Oxytocin |
2.2 |
TCEP |
A |
1:5 |
1 |
6 |
1 h, 37 C |
|||||||
|
zur Untersuchung der Spezifität der Modifizierungsreaktion: |
||||||||||||||
|
Oxytocin |
2.4.1 |
ME |
B & C |
1:50 |
2 |
100 |
2 h, 37 C |
|||||||
|
DTT |
B & C |
1:10 |
2 |
25 |
2 h, 37 C |
|||||||||
|
TCEP |
B & C |
1:5 |
2 |
5 |
2 h, 37 C |
|||||||||
|
für die Umsetzung mit den synthetisierten Derivaten der Arsonigen Säuren: |
||||||||||||||
|
Vasopressin |
2.3.2 |
DTT |
B |
1:5.4 |
2 |
10 |
1 h, 37 C |
|||||||
|
TCEP |
C |
1:5.4 |
2 |
10 |
1 h, 37 C |
|||||||||
|
zur Darstellung von teilreduziertem BPTI |
||||||||||||||
|
BPTI |
2.6.2 |
TCEP |
A |
1:60 |
1 |
18 |
2-4 h, 37 C |
|||||||
|
zur Darstellung von vollständig reduziertem BPTI, Insulin und rhM-CSF |
||||||||||||||
|
BPTI |
2.6.2 |
TCEP |
D |
1:60 |
1 |
18 |
2 h, 37 C |
|||||||
|
Insulin |
2.6.2 |
TCEP |
E |
1:60 |
2 |
21 |
7 h, 37 C |
|||||||
|
rhM-CSF |
2.7.1 |
TCEP |
A |
1:180 |
2.8 |
12 |
5 h, 37 C |
|||||||
3.4.2 Alkylierung von Cysteinylthiolgruppen mit Iodacetamid und 4-Vinylpyridin
Die für die Peptide (Oxytocin, Vasopressin) und Proteine (BPTI, Insulin, rhM-CSF) durchgeführten Alkylierungsreaktionen sollten die vollständige Reduktion (siehe 3.4.1) bestätigen. Dabei wurde ein 2.2-facher molarer Überschuß an Iodacetamid (IAA) oder 4-Vinylpyridin (VP) bezogen auf das jeweils eingesetzte Reduktionsmittel gewählt. Die Alkylierungsreaktionen erfolgten stets in dem für die Reduktion verwendeten Puffer (siehe Tab. 16). Alkylierungen mit VP wurden für 30 min bei 37 C durchgeführt. Alkylierungen mit IAA wurden für 1 h bei 25 C im Dunkeln durchgeführt, wobei der pH-Wert nach 5 und 20 min überprüft und gegebenenfalls mit 2.5% wäßriger Ammoniaklösung eingestellt wurde. Die Alkylierungsreaktionen für IAA und VP wurden durch Absenken des pH-Werts auf pH 3 mit 3-5 Tropfen Eisessig beendet.
Anmerkungen:
- Die Reaktionsbedingungen für die Alkylierung der rhM-CSF Faltungsintermediate sind in 3.8 beschrieben.
- Die UV-spektroskopischen Untersuchungen zu den Reaktionen von Oxytocin mit IAA und VP in Anwesenheit von MEL sind in 3.4.3 (d) beschrieben.
3.4.3 Modifizierung von Biscysteinylthiolgruppen mit Arsonigen Säuren
Folgende Puffer wurden für die Modifizierungsreaktionen verwendet:
|
Allgemeines Durchführungsschema der UV-spektroskopischen Untersuchung der Modifizierungsreaktion von Bisthiolen mit MEL:
Alle Umsetzungen erfolgten in Quarzglasküvetten (siehe 3.3). Es wurden außer den in Abschnitt 3.3.2 aufgeführten Stammlösungen an MEL entsprechende Puffer mit gleicher MEL-Konzentration mit zusätzlich 6 M Harnstoff verwendet. Je nach verwendetem Puffersystem (Nullabgleich des Photometers), in dem die Modifizierungsreaktion durchgeführt werden sollte, wurde die entsprechende MEL-Stammlösung mit dem entsprechenden Puffer so verdünnt, daß nach Zugabe der (meist) reduzierten Proteinlösung ein Gesamtvolumen von 1 ml resultierte. Die so verdünnte MEL-Lösung wurde in der UV-Küvette vorgelegt. Der zeitliche Verlauf der Absorptionswerte bei 267 nm und 299 nm wurde vor der Zugabe des zu modifizierenden Proteins gestartet. Nach ca. 60 s wurde das UV-Photometer geöffnet und die entsprechende Proteinlösung zur MEL-Lösung pipettiert, mit einem Mikrospatel umgerührt und das UV-Photometer wieder geschlossen. Dieser Vorgang dauerte durchschnittlich 15-30 s. Nach beendeter Modifizierung wurden der zeitliche Verlauf der Absorptionsdifferenz berechnet.
(a) Umsetzungen SP-C(1-14) mit PAA, PYR, A-PAA und MEL
Der Anteil an organischem Lösungsmittel sollte während der Modifizierungsreaktion 5% (v/v) sein. 440µg lyophilisiertes SP-C(1-14) wurde in einer Konzentration von 1µg/µl in Puffer A gelöst; diese Lösung wurde dann in Aliquote à 90 µg (58 nmol) unterteilt. Für die Umsetzung mit PAA wurde ein 90 µg Aliquot mit 375 µl Puffer A verdünnt (csp-c(1-14) = 0.18 µg/µl) und mit 25 µl einer 10 mM PAA-Lösung in DMSO versetzt. Die Umsetzung mit PAA erfolgte im molaren Verhältnis SP-C(1-14) / PAA von 1:4.3 bei einer PAA-Konzentration von 0.5 mM. Drei weitere 90 µg SP-C(1-14) Aliquote wurden jeweils mit 290 µl Puffer A verdünnt (csp-c(1-14) = 0.23 µg/µl) und mit 13 µl einer 100 mM PYR-, A-PAA- und MEL-Lösung in MeOH / H2O 9:1 versetzt. Das molare Verhältnis SP-C(1-14) / Arsoniger Säure betrug jeweils 1:23 bei einer Konzentration von 3.3 mM PYR, A-PAA bzw. MEL. Alle Lösungen, bis auf die von A-PAA, wurden frisch zubereitet. Die Lösungen wurden nach 45 min bei 25 C mit 500 µl 0.1% TFA angesäuert und über SepPak-RP-C18-Kartuschen gereinigt (siehe 3.6.1).
(b) Umsetzungen von reduziertem Vasopressin und Oxytocin mit PAA, PYR, A-PAA und MEL
Quantitative Umsetzung von vollständig reduziertem Vasopressin:
Je vier 400 µg (369 nmol) Aliquote Vasopressin (Vas) wurden mit DTT bzw. TCEP reduziert (siehe 3.1.1). Die Modifizierungsreaktion erfolgte in einem molaren Verhältnis Vasopressin / Reduktionsmittel / Arsonige Säure = 1:5.4:13 in Puffer A (TCEP-Reduktion) und Puffer B (DTT-Reduktion). Für die Umsetzung mit PAA wurde eine Lösung von 400 µg reduziertem Vasopressin in 384 µl Puffer mit 476 µl 10 mM PAA in DMSO versetzt. Während der Modifizierungsreaktion betrug der Anteil an DMSO 55% (v/v), die einzelnen Konzentrationen waren: cvas = 0.23 µg/µl, credm. = 2.3 mM und cpaa = 5.5 mM. Für die Umsetzungen mit PYR, A-PAA und MEL wurden jeweils 400 µg Aliquote an reduziertem Vasopressin in 384 µl Puffer A bzw. B mit je 16 µl der frisch zubereiteten 300 mM PYR-, A-PAA- und MEL-Stammlösungen in MeOH / H2O 9:1 versetzt. Der Anteil an MeOH während der Modifizierungsreaktion betrug 4% (v/v), die einzelnen Konzentrationen waren: cvas = 1 µg/µl, credm. = 5 mM und cPYR / A-PAA / MEL = 12 mM. Nach 1 h bei 37 C wurden die aus den Lösungen präzipitierten Niederschläge abzentrifugiert (1 min, 10000 g), die Überstände abgetrennt und bei -20 C aufbewahrt.
Umsetzung vom partiell reduziertem Vasopressin:
1 mg (992 nmol) Vasopressin wurden bei einer Konzentration cvas = 1 µg/µl mit DTT im molaren Verhältnis 1:5 in Puffer B reduziert (cdtt = 4.5 mM). Zu bestimmten Zeiten wurden 10 µg Aliquote entnommen, lyophilisiert und FAB-massenspektrometrisch charakterisiert (Matrix: 4% TFA / Glycerin). Beim Verhältnis an oxidiertem zu reduziertem Vasopressin von ca. 1:2 wurde die Reduktion durch 10-faches Verdünnen mit 50 mM NH4OAc pH 3.5 gestoppt. Die Lösung (cvas = 0.1 µg/µl, pH 4) wurde mit 8 µl einer 300 mM PYR-Lösung in MeOH / H2O 9:1 versetzt und 1h bei 37 C inkubiert. Die Modifizierung mit PYR erfolgte im molaren Verhältnis von Vasopressin / DTT / PYR 1:5:6 mit einem Anteil an organischem Lösungsmittel von < 2% (v/v). Das Lyophilisat der Reaktionslösung wurde in 0.1% TFA gelöst, MALDI-massenspektrometrisch charakterisiert und ein Aliquot (ca. 20 µg) auf eine C18-RP-HPLC-Säule aufgetragen (siehe 3.6.3). Die erhaltenen Fraktionen wurden vor dem Lyophilisieren mittels MALDI-MS charakterisiert und nach Lyophilisieren in 2% AcOH / MeOH 9:1 gelöst (ca. 0.1 µg/µl) und mittels ESI-MS und ESI-FT-ICR-MS charakterisiert.
Zweifachmodifizierungen:
100 µg (100 nmol) Aliquote an Oxytocin wurde nach Reduktion (siehe 3.4.1) bei pH 4 (Puffer A) und pH 8 (Puffer B) in folgenden molaren Verhältnissen mit MEL umgesetzt: OT / ME / MEL 1:50:110; OT / DTT / MEL 1:10:22 und OT / TCEP / MEL 1:5:11. Analog erfolgte die Umsetzung mit PAA, PYR und A-PAA. Es wurden folgende Stammlösungen verwendet: 10 mM PAA in DMSO und je 300 mM PYR, A-PAA und MEL in MeOH / H2O 9:1. Nach 1 h bei 37 C wurde der pH durch Verdünnen mit 0.1% TFA auf zwei eingestellt und die Reaktionslösungen ohne weitere Reinigung lyophilisiert. Die Lyophilisate wurden nach Lösen mit 4% TFA (cot 2 µg/µl) mittels MALDI-MS charakterisiert.
(c) In situ-Modifizierung von SP-C mit MEL auf dem PD-Probentarget
1.5 µg (0.42 nmol) SP-C wurden auf dem PD-Probentarget (siehe 3.7.3) mit MEL im molaren Verhältnis 1:20 umgesetzt. Dazu wurden zweimal 5 µl einer Lösung von SP-C (0.15 µg/µl) in Puffer C auf das PD-Probentarget aufgetragen und nach dem Trocknen mit 1.7 µl einer 5 mM MEL-Lösung in Puffer C versetzt. Das Target wurde 20 min unter einem Deckgläschen in Wasserdampfatmosphäre bei 37 C stehen gelassen. Nach Trocknung wurden anschließend überschüssiges MEL durch Waschen mit ca. 50 µl 0.1% TFA entfernt. Nach erneuter Trocknung wurde der Probenträger direkt zur PD-MS-Analyse eingesetzt.
(d) UV-spektroskopische Untersuchung der Reaktion von Oxytocin mit MEL und in situ-Reduktion mit TCEP
225 µl 0.2 mM MEL (45 nmol) in Puffer B wurden in der UV-Küvette mit 745 µl Puffer B verdünnt, die Absorptionsmessung gestartet und nach ca. 60 s 30 µg Oxytocin (30 nmol) in 10 mM NH4HCO3 pH 8.2 zugegeben. Zu bestimmten Zeitpunkten (siehe 2.3.2) wurden 1.5, 5.0 bzw. 10 µl 100 mM TCEP in H2O zu dieser Lösung pipettiert und der pH-Wert nach Mischen kontrolliert. Aliquote der Reaktionslösung wurden MALDI-massenspektrometrisch charakterisiert (siehe 3.7.5). Analog wurde die Reaktion in Puffer A durchgeführt.
(e) UV-spektroskopische Untersuchung des Reaktionsverlaufs von reduziertem Oxytocin mit IAA oder VP in Gegenwart von MEL
200 µl 0.2 mM MEL (40 nmol) in Puffer B wurden in der UV-Küvette mit 770 µl Puffer B verdünnt, die Absorptionsmessung gestartet und nach ca. 60 s 30 µg reduziertes Oxytocin (30 nmol) (siehe 3.4.1) zugegeben. Zur Kontrolle wurde der identische Reaktionsansatz ohne MEL in einem Eppendorf-Reaktionsgefäß vorbereitet. Zu bestimmten Zeitpunkten (siehe 2.5) wurden zur Lösung in der UV-Küvette und zur Kontrolle 2 und 10 µl einer 100 mM VP-Lösung in H2O zugegeben. Aliquote der Reaktionslösung und der Kontrolle wurden mittels MALDI-MS charakterisiert (siehe 3.7.5). Analog wurde die Reaktion in Puffer D durchgeführt.
Die Umsetzung in Puffer D wurde abgewandelt und erweitert: Statt VP wurden 13 µl einer 100 mM IAA-Lösung in H2O zugegeben und der pH-Wert kontrolliert. Nach 30 min Inkubationszeit mit IAA wurden 5 µl einer 100 mM DTT-Lösung in H2O zugegeben und nach 50 sec und 3 h Aliquote MALDI-massenspektrometrisch (siehe 3.7.5) charakterisiert.
(f) UV-spektroskopische Untersuchung der Reaktion von reduziertem Oxytocin mit MEL in Gegenwart von GSH
200 µl 0.2 mM MEL (40 nmol) in Puffer D (inklusive 0.7 M Harnstoff) wurden in der UV-Küvette mit 684 µl des entsprechenden Puffers verdünnt, die Absorptionsmessung gestartet und nach ca. 60 s 39 µg GSH (125 nmol) in Puffer D (inklusive 0.7 M Harnstoff) zugegeben. Nach einer Reaktionszeit von ca. 2400 s wurden 30 µg reduziertes Oxytocin (30 nmol) (siehe 3.4.1) zugegeben. Aliquote der Reaktionslösung wurden mittels MALDI-MS charakterisiert (siehe 3.7.5).
(g) UV-spektroskopische Untersuchung des Reaktionsverlaufs von SP-C und reduziertem BPTI, Insulin und rhM-CSF mit MEL
100 µl 0.2 mM MEL (20 nmol) in Puffer C wurden in der UV-Küvette mit 250 µl Puffer C verdünnt und die Absorptionsmessung gestartet und nach ca. 60 s 88 µg (24.8 nmol) synthetisches SP-C zugegeben.
200 µl 0.2 mM MEL (40 nmol) in Puffer E wurden in der UV-Küvette mit 735 µl Puffer E verdünnt, die Absorptionsmessung gestartet und nach ca. 60 sec. 65 µg (10 nmol) teilreduziertes (siehe 3.4.1)BPTI (1 µg/µl) zugegeben.
200 µl 0.2 mM MEL (40 nmol) in Puffer D wurden in der UV-Küvette mit 735 µl Puffer D (inklusive 6 M Harnstoff) verdünnt, die Absorptionsmessung gestartet und nach ca. 60 s 65 µg (10 nmol) vollständig reduziertes (siehe 3.4.1) BPTI (1 µg/µl) in zugegeben.
100 µl 0.4 mM MEL (40 nmol) in 10 mM Puffer F wurden in der UV-Küvette mit 866 µl Puffer F (inklusive 6 M Harnstoff) verdünnt, die Absorptionsmessung gestartet und nach ca. 60 s 69 µg (12 nmol) vollständig reduziertes (siehe 3.4.1) Insulin (2 µg/µl) zugegeben.
200 µl 0.2 mM MEL (40 nmol) in Puffer E wurden in der UV-Küvette mit 742 µl Puffer E (inklusive 6 M Harnstoff) verdünnt, die Absorptionsmessung gestartet und nach ca. 60 s 163 µg (3.2 nmol) vollständig reduziertes (siehe 3.4.1) rhM-CSF-Monomer (2.8 µg/µl) in zugegeben.
Alle Aliquote aus den Reaktionslösungen wurden ohne weitere Reinigung mittels MALDI-MS charakterisiert (siehe 3.7.5).
3.4.4 Proteolytische Abbaureaktionen
Alle proteolytischen Abbaureaktionen wurden mit HPLC-gereinigtem (siehe 3.6.3) MEL-modifiziertem Vasopressin (Vas-MEL) und HPLC-gereingter MEL-modifizierter B-Kette des Insulins (Ins-B-MEL) durchgeführt. Die entsprechenden HPLC-Fraktionen wurden in ca. 5 µg Aliquote unterteilt und lyophilisiert.
Zwei Aliquote einer Lösung von ca. 5 µg Vas-MEL in 10 µl 50 mM NaH2PO4 pH 8.5 wurden mit 1µl einer 1 µg/µl Trypsin-Lösung (TPCK-behandelt) bzw. mit 1 µl einer 1 µg/µl -Chymotrypsin-Lösung versetzt und bei 25 C inkubiert. Nach 15, 30, 45, 60 min und nach 3 h wurden 0.5 µl der Reaktionslösungen entnommen und MALDI-massenspektrometrisch charakterisiert.
Drei Aliquote einer Lösung von ca. 5 µg Ins-B-MEL wurden in 5 µl 1 M Harnstoff, 50 mM NaH2PO4 pH 8.5 gelöst und jeweils mit 1 µl einer 1 µg/µl Trypsin-Lösung (TPCK-behandelt), 1 µg/µl -Chymotrypsin-Lösung und 1 µg/µl V8-Protease-Lösung versetzt. Ein weiteres 5 µg Aliquot Ins-B-MEl wurde in 5 µl 1 M Harnstoff, 50 mM NaH2PO4 pH 4.0 gelöst und mit 1 µl einer 1 µg/µl V8-Protease-Lösung versetzt. Die proteolytischen Abbaureaktionen erfolgten bei 25 C. Nach 30, 60 und 100 min wurden 0.5 µl der Reaktionslösungen entnommen und MALDI-massenspektrometrisch charakterisiert.
3.4.5 Bestimmung der Thiolkonzentration mit Ellmans Reagenz
Für die UV-spektroskopische Untersuchung der Reaktion von MEL mit Dithiolen wurden Stammlösungen von je 10 mM DMP und DTT in 10 mM NH4HCO3 pH 8.2 hergestellt. Die Stammlösungen wurden 1:10 mit 10 mM NH4HCO3 pH 8.2 verdünnt und die Verdünnungen bis zur Verwendung auf Eis gestellt. 2,2'-Dinitro-5,5'dithiodibenzoesäure (DTNB) wurde als 2 mM Lösung in 10 mM NH4HCO3 pH 8.2 verwendet. Je 10, 20, 30 µl der zu untersuchenden Thiol-Lösungen wurden im Eppendorf-Reaktionsgefäß (1.5 ml) mit NH4HCO3-Puffer (10 mM, pH 8.2) auf 950 µl aufgefüllt, gut gemischt und mit 50 µl der DTNB-Lösung versetzt. Nach sorgfältigem Mischen und ein-minütiger Inkubation bei 25 °C wurde die Absorption bei 412 nm gegen die Referenz (950 µl NH4HCO3-Puffer (10 mM, pH 8.2) und 50 µl der DTNB-Lösung) gemessen. Als Wert für den Extinktionskoeffizenten des entstandenen 2-Nitro-5-thiolatbenzoatdianions wurde = 13600 cm-1 M-1 verwendet [43]. Die so erhaltenen Thiolkonzentration wurden gemittelt. Es zeigte sich, daß die Thiolkonzentration von DTT bei 4C über vier Stunden konstant blieb, während für DMP bei 4C innerhalb von 90 min nur noch 75% der ursprünglichen Thiolkonzentration vorlag.
3.5 Stabilitätsuntersuchungen
Zur Überprüfung der Stabilität von MEL-modifiziertem Vasopressin (Vas-MEL) wurden Aliquote à ca. 10 µg (7.3 nmol) der HPLC-gereingten Fraktionen (siehe 3.6.3) lyophilisiert. Die einzelnen Aliquote wurden in den entsprechenden Puffer (siehe 2.4.2) gelöst und bei 25 C inkubiert. Ein weiteres Aliquot wurde mit 20 µl einer Lösung bestehend aus 4µl 5 M BrCN in ACN und 16 µl 0.1% TFA / 0.1% TFA in ACN 6:4 (pH 2) versetzt und ebenfalls bei 25 C inkubiert. Nach 30 min, 1 h, 3h, 8 h und 25 h wurden 0.5 µl Aliquote der Reaktionslösungen entnommen und MALDI-massenspektrometrisch charakterisiert.
In einer analog durchgeführten Versuchsreihe wurden Aliquote mit je ca. 5 µg (3.7 nmol) Vas-MEL in den entsprechenden Puffer gelöst, mit 1 µl einer 100 mM DTT-Lösung (100 nmol) in H2O versetzt und bei 25 C inkubiert. Nach 15, 30 und 60 min wurden 0.5 µl Aliquote der Reaktionslösungen entnommen und MALDI-massenspektrometrisch charakterisiert.
3.6 Proteinreinigung und Proteinbestimmung
3.6.1 SepPak-C18-RP-Kartuschen
Das mit MEL modifizierte SP-C(1-14) wurde über C18-RP-Kartuschen (Water-Millipore, Typ: SepPak) gereinigt. Die Kartuschen wurden zunächst mit je 10 ml MeOH und 0.1% TFA equilibriert. Das Rohprodukt wurde dann in 1 ml 0.1% TFA aufgetragen. Mit 10 ml 0.1% TFA wurden zuerst die Reagenzienüberschüsse, mit weiteren 10 ml 0.1% TFA / ACN 20:80 schließlich das Produkt eluiert. Die Produktfraktionen wurden vereinigt und lyophilisiert.
3.6.2 Ultrafiltration (Microcon)
Das von der Fa. Chiron zur Verfügung gestellte rhM-CSF und die mit IAA alkylierten bzw. MEL-modifizierten rhM-CSF-Faltungsintermediate wurden durch Ultrafiltration gereinigt. Die Probenlösungen (200-500 µl) wurden in die Microkonzentratoren Microcon 30 (30 kD Molekularauschlußgewicht, Fa. Amicon) überführt und durch Zentrifugation (10 min, 10000 g) aufkonzentriert (ca. 100 µl). Nach zwei Waschschritten mit den jeweils verwendeten Puffern wurde der Retentatbehälter um 180 gedreht und das Retentat (ca. 100 µl) in ein separates Mikroreaktionsgefäß durch Zentrifugation (3 min, 1000 g) überführt.
3.6.3 Hochdruckflüssigkeitschromatographie
Zur Reinigung von Peptid- und Proteinderivaten wurde die Reversed-Phase-Hochdruckflüssigkeitschromatographie eingesetzt. Es wurde ein modifiziertes HPLC-System (Waters Millipore) verwendet, das mit einem binären Gradientensystem aus zwei Präzisionshochdruckpumpen (Waters M510, Waters M45) ausgestattet war. Zur Detektion wurde ein Multiwellenlängen-UV-Detektor (Waters 490E) verwendet. Die Untersuchungen wurden bei Raumtemperatur und mit einer Flußrate von 1 ml/min durchgeführt. Die Probenaufgabe erfolgte mit einer Mikroliterspritze mit Hilfe eines drucklosen Injektorsystem. Die HPLC-Untersuchungen wurden mit einer 250 x 4 mm analytischen Säule (C18-RP, 300 Å Porenweite, 7 µm Partikelgröße, Vydac) durchgeführt. Alle Lösungsmittel wurden vor Verwendung durch 0.45 µm Nylon-Membranfilter (NL17, Schleicher & Schuell) filtriert und anschließend 20 min im Ultraschallbad (Transsonic 570, Elma) unter Wasserstrahlvakuum entgast. Zur Reinigung von MEL- und PYR-modifiziertem Vasopressin wurde ein Gradientensystem aus 0.1% (v/v) wäßriger TFA (A) und 0.07% (v/v) TFA in Acetonitril (B) verwendet. Folgender Lösungsmittelgradient wurde angewendet: 0-5 min konstant bei 5% B, von 5-35 min linear bis 65% B, von 35-40 min linear bis 98% B, bis 45 min konstant bei 98% B, von 45-50 min linear bis 5% B und bis 55 min konstant bei 5% B. Zur Reinigung von MEL-modifiziertem Insulin wurde ein Gradientensystem aus 0.1% (v/v) wäßriger TFA (A) und 0.1% (v/v) TFA in Acetonitril (B) verwendet. Der Gradient war von 0-5 min konstant bei 10% B, von 5-10 min linear bis 20% B, von 10-30 min linear bis 40% B, von 30-40 min linear bis 90% B, bis 43 min konstant bei 90% B, von 43-52 min linear bis 10% B und bis 60 min konstant bei 10% B. Es wurden jeweils die Reaktionslösungen nach der Modifizierungsreaktion ohne weitere Behandlung aufgetragen (80-120 µg). Die Fraktionen wurden in Eppendorf-Reaktionsgefäßen isoliert und MALDI-massenspektrometrisch charakterisiert.
3.6.4 Polyacrylamid-Gelelektrophorese (SDS-PAGE)
Für die Polyacrylamid-Gelelektrophorese wurde die Mini-Protean II Anlage von Biorad benutzt. Die Mengenangaben beziehen sich auf das Gießen zweier 0.4 mm dicker Gele. Zur Molekulargewichtsbestimmung wurde der Molekulargewichtsmarker LMW (Pharmacia) mit folgenden Proteinen verwendet: Phosphorylase B (94 kD), Albumin (67 kD), Ovalbumin (43 kD), Carbonic Anhydrase (30 kD), Trypsin-Inhibitor (20.1 kD) und -Lactalbumin (14.4 kD).
(a) SDS-PAGE nach Laemmli
Es wurden Polyacrylamid-Gele mit 12%igem Trenngel und 5%igem Sammelgel nach Laemmli verwendet, die sich insbesondere für die Analyse größerer Proteine über 20 kD eignen [156]. Zuerst wurde das Trenngel gegossen und mit MilliQ überschichtet. Nach der Polymerisation wurde das Wasser abgegossen und das Sammelgel mit eingesetztem Probenkamm polymerisiert. Alle verwendeten Lösungen wurden mit MilliQ hergestellt (siehe Tab. 17).
Tab. 17: Zusammensetzung der Polymerisationslösungen für die SDS-PAGE nach Laemmli.
|
verwendeteLösungen |
Zusammensetzung |
Sammelgel 5 % |
Trenngel12 % |
|
4 x Sammelgelpuffer |
0.5 M Tris/HCl, 0.4% (w/v) SDS, pH 6.8 |
2.5 ml |
- |
|
4 x Trenngelpuffer |
1.5 M Tris/HCl, 0.4% (w/v) SDS, pH 8.8 |
- |
6 ml |
|
MilliQ |
- |
5.8 ml |
6 ml |
|
Acrylamid-BIS |
30% (w/v) Acrylamid, 0.8% (w/v) Methylenbisacrylamid |
1.7 ml |
9.6 ml |
|
APS 10% |
10% (w/v) Ammoniumperoxodisulfat |
85 µl |
125 µl |
|
TEMED |
N,N,N',N'-Tetramethylethylendiamin (100%) |
20 µl |
20 µl |
Aufschlußpuffer (nicht reduzierend): 4% (w/v) SDS, 12 % (w/v) Glycin, 50 mM Tris/HCl,
0.02% (w/v) Coomassie Brilliant Blue G-250, pH 6.8
Die untersuchten Proben wurden 1:1 mit dem Aufschlußpuffer verdünnt und unmittelbar vor der Aufgabe auf das Gel 5 min bei 95 C denaturiert. Die Elektrophorese wurde mit konstant 30 mA durchgeführt, bis die Farbstoffbanden das Ende des Trenngels erreicht hatten.
(b) Coomassie-Blau-Färbung
Für die Coomassie-Blau-Färbung wurden folgende Lösungen verwendet:
Fixierlösung: 50% (v/v) MeOH, 10% (v/v) AcOH, 40% MilliQ
Färbelösung: 10% (v/v) AcOH, 0.05% Coomassie Brilliant Blue G-250
Entfärbelösung: 10% (v/v) AcOH
Die Gele wurden unter leichtem Schwenken 30 min in der Fixierlösung fixiert, anschließend mindestens 2 h gefärbt und dann etwa 2-4 h unter mehrfachem Wechsel der Entfärbelösung entfärbt. Die Gele wurden dann auf feuchtes Chromatographiepapier übertragen und 1 h bei 50 C unter Wasserstrahlvakuum in einem Aldo-Xer-Geltrockner (Schütt) getrocknet.
3.6.5 Bradfordassay zur Bestimmung der Proteinkonzentration
Die Proteinkonzentrationsbestimmungen wurden nach der von Bradford entwickelten "dye binding method" [157] unter Verwendung von 96-well Mikrotiterplatten durchgeführt. Die Methode beruht auf der Verschiebung des Absorptionsmaximums des Farbstoffs Coomassie Brilliant Blue G-250 von 465 nach 595 nm durch Bindung an das Protein. Die Proteinkonzentration wurde mit Hilfe einer parallel mitbestimmten Eichkurve von Rinderserum-Albumin (BSA) bekannter Konzentration ermittelt. Zur Aufstellung einer Eichkurve wurde eine Konzentrationsreihe von BSA mit sechs Konzentrationen zwischen 0.0156 und 0.5 µg/µl im gleichen Puffer wie das zu bestimmende rhM-CSF gemessen. Jeweils 20 µl der rhM-CSF-Lösung (siehe 3.4.1) wurden mit 180 µl Bradfordreagenz in eine Vertiefung der ELISA-Platte (Greiner 655101) gemischt. Fünf Minuten nach Zugabe des Bradfordreagenz wurde die Extinktion der Proben mittels eines Anthos HTL2-ELISA-Readers (Anthos Labtec) bei 595 bestimmt. Das Bradfordreagenz wurde aus 3 ml Stammlösung (50 mg Coomassie Brilliant Blue G-250, 50 ml 95%iges Ethanol, 100 ml 88%ige Phosphorsäure) und 17 ml MilliQ jeweils frisch gemischt.
3.7 Analytische Charakterisierungsmethoden
3.7.1 Elektronen-Ionisations-Massenspektrometrie
Die EI-massenspektrometrischen Untersuchungen wurden mit einem doppelfokusierenden Massenspektrometer (Finnigan MAT 312 ) durchgeführt. Die Proben wurden durch Direktverdampfung in die Gasphase überführt. Die Ionisation erfolgte durch Beschuß der gasförmigen Probe mit Elektronen (70 eV). Die Quellentemperatur wurde konstant bei 200 C gehalten. Die Messungen erfolgten bei einer Beschleunigungsspannung von 3-6 kV, einer Scangeschwindigkeit von 14 sec / Dekade, und einer Auflösung von etwa 800. Zur Massenkalibrierung diente das Massenspektrum von (hochviskosem) Perfluorkerosin, das Signale bis etwa m/z 850 lieferte.
3.7.2 Fast-Atom-Bombardment-Massenspektrometrie
Die FAB-massenspektrometrischen Untersuchungen wurden mit einem modifizierten doppelfokusierenden Massenspektrometer (Finnigan MAT 312/ AMD 5000) mit Nier-Johnson Geometrie durchgeführt. Als Primärionenquelle wurde eine 20 kV Cs+-Thermoionenquelle bei einer Heizstromstärke von 2 A und einer Emission von ca. 2-5 µA verwendet. Aufgrund des Hochfeldmagneten (AMD, Beckelen) ist ein Massenbereich bis 2100 AMU bei 6 kV und 5000 AMU bei 3 kV erfaßbar. Zur Spektrenaufnahme wurde ein Direkteinlaßsystem mit einem Probentarget verwendet, das aus einer abgeschrägten Stahlfläche (ca. 2 mm2 Oberfläche) in einem Winkel von 60 zur Primärionenquelle bestand.
Die Proben wurden in folgenden Lösungsmittelgemischen gelöst: DMSO / Eisessig / Glycerin 1:1:1; Glycerin / 1N HCl 1:1; DMSO / Eisessig / 3-Nitrobenzylalkohol 1:1:1; DMSO / Eisessig / 2-Nitrophenyl-n-octylether 1:1:1. Anschließend wurden 1-2 µl dieser Lösungen auf den FAB-Probenträger überführt und anschließend mit einer Direkteinlaßschubstange in die Ionenquelle eingeführt. Die Messungen erfolgten bei einer Beschleunigungsspannung von 3-6 kV, einer Scangeschwindigkeit von 14 sec / Dekade, einer Auflösung von etwa 2500 und einer Betriebsspannung des Sekundärionen-Vervielfachers (SEV) von 2-2.4 kV. Zur Massenkalibrierung diente das Clusterionenspektrum einer CsJ / Glycerin-Lösung, die regelmäßige Serien von Clusterionen im Bereich von 100-2500 amu lieferte [158].
3.7.3 252Cf-Plasmadesorptions- Massenspektrometrie
Die PD-MS Untersuchungen wurden mit einem BIO ION 20 K Flugzeit-Massenspektrometer (BIO ION NORDIC AB, Upsalla, Schweden) durchgeführt. Die Primärionenquelle bestand aus einer 10 µCi 252Cf-Quelle in einer versiegelten Gold-Nickel-Folie, deren Zerfallsfragmente (ca. 1000-1500 Nuklide pro Sekunde, z.B. 142Ba18+, ca. 80 MeV; 106Tc22+, ca. 100 MeV) die Desorption der Sekundärionen von der Oberfläche einer mit Nitrocellulose besprühten Aluminiumfolie induzierten. Die desorbierten Molekülionen wurden durch eine Spannung von 16 kV beschleunigt und ihre Flugzeit nach 13.9 cm Flugstrecke durch einen Stop/Zeitdetektor gemessen. Die Zeitmessung erfolgte durch einen "time-to-digital-converter" (TDC) bei einer maximalen Zeitauflösung von 1 ns / Meßkanal. Zur Aufnahme wurden die Sekundärspektren von 5-40 Millionen Zerfallsereignissen akkumuliert. Das resultierende Flugzeitspektrum wurde mit dem "DAS" Datensystem (BIO ION) auf einem PDP 11/73 Computer (Digital Equipment Corp.) durch Zweipunktkalibrierung mit den Referenzmassen von H+ und NO+ (aus dem Spektrenuntergrund der Nitrocellulose) in Massenspektren überführt.
Als Probenträger wurden Messingringe mit einem Durchmesser von 1 cm verwendet, die mit einer doppelseitig aluminiumbedampften Polyethylenterephthalatfolie (Mylarfolie, Alexander Vacuum Research Inc., Greenfield, USA) bespannt wurde. Zur Probenadsorption wurden die Träger unmittelbar vor der Analyse mit Hilfe eines Elektrospray-Geräts (BIO ION) durch Aufsprühen einer Lösung von Nitrocellulose (BA 83, Schleicher & Schuell, Dassel) von 2 µg/µl in Aceton und einer Flußrate von 15 µl/s mit 100-200 µg Nitrocellulose beschichtet. Die Probenlösungen (1-5 µl, Substanzmenge 1-5 nmol) wurden mit einer Mikroliterpipette auf dem Nitrocellulose-Träger pipettiert und dieser nach etwa 5 min durch die "spin-drying"-Technik getrocknet [159].
3.7.4 Elektrospray- Massenspektrometrie
Die ES-MS Untersuchungen wurden mit einem Vestec 201A Single-Quadrupol-Massenspektrometer (Vestec Corp., Houston, USA) mit einem Massenbereich bis 2000 amu und Einheitsauflösung durchgeführt. Das Elektrospray wurde durch Anlegen einer Spannung von 1.8-2.3 kV an der Nadelspitze und 200 V an der Nozzle erzeugt. Die Messungen wurden wie beschrieben bei verschiedenen Repellerspannungen (5-100 V) und einer Repellerblocktemperatur von 200 C durchgeführt. Die Spray-Kammer wurde zwischen 40 und 65 C thermostatisiert. Die Massenkalibrierung erfolgte anhand der 8-, 9- und 10-fach geladenen Molekülionen des Lysozyms bzw. der im Massenbereich < 1000 amu anhand der Molekülionen des oligomeren Pyridin-3-arsonigsäureanhydrids (siehe Abb. 26). Die massenspektrometrischen Analysen wurden mit Hilfe eines Datensystems (Vector 2, Teknivent, Maryland, USA) aufgezeichnet und ausgewertet.
Zur Probenpräparation wurden die Proben in 2% Essigsäure / Methanol 9:1 für die beschriebenen Konzentrationen gelöst. 100 µl dieser Lösung wurden mittels eines drucklosen Ventils auf eine ca. 40 cm lange Fused Silica-Kapillare (Innendurchmesser 50 µm) überführt und mit Hilfe eines Pumpsystem (Harward Apparatus 44) bei einer Flußrate von 2-3 µl in die Ionenquelle gepumpt.
3.7.5 Matrixunterstützte-Laserdesorptions-/Ionisations- Massenspektrometrie
Die Spektrenaufnahme erfolgte mit einem Bruker-Biflex MALDI-TOF Instrument der Firma Bruker-Franzen, Bremen mit einer SCOUT-Quelle. Die Spektrenaufnahme von Peptiden/Proteinen mit einem MW < 10 kD erfolgte bei Beschleunigungsspannungen von 15-20 kV und der zur Desorption minimal notwendigen Laserleistung (50-80%). Diese wurde während der Messung mit dem Attenuator eingestellt und anschließend nicht mehr verändert. Proteine mit einem MW > 10 kD wurden bei einer Beschleunigungsspannung von 25 kV gemessen. Die interne Kalibrierung erfolgte bei Proben mit MW < 2000 durch Matrixionen und SP-C(1-14), bei MW < 7000 durch Insulin bzw. BPTI (jeweils der Matrix beigemengt) und bei höhermolekularen Proben mit MW < 50 kD durch Trypsinogen (bis 25 kD), TU und rhM-CSF-Homodimer. Intensive Matrixsignale, die insbesondere bei Proben mit MW > 5000 die Detektionsempfindlichkeit negativ beeinträchtigen, wurden durch Ausblendung der Matrixionen mit einem einstellbaren Deflektionsimpuls (3.5-4.5 µs) eliminiert. Die Auswertung der Massenspektren erfolgte mittels des Datenauswertsystems XMASS (Bruker-Franzen GmbH) auf einer Sparc 1-Workstation (SUN).
Zur Probenpräparation wurde gesättigte -Cyano-4-hydroxyzimtsäure (HCCA)-Lösung in ACN / 0.1% TFA 2:1 als Standard-Matrixlösung verwendet. Für die Stabilitätsuntersuchungen von HPLC-gereinigtem, MEL-modifizertem Vasopressin wurden weiterhin die Matrizes 6-Aza-2-thiothymin (ATT) in Aceton / H2O 9:1 (10 mg/ml) und 2,6-Dihydroxyacetophenon (DHAP) als gesättigte Lösung in Aceton verwendet. Alle untersuchten Peptide und Peptidderivate (0.05 bis 1µg) wurden in Abwandlung der Dried-Droplet-Methode im Volumenverhältnis 1:1 mit der Matrixlösung auf dem Edelstahlprobenträger (Target) vermischt und bei Raumtemperatur getrocknet. Für die untersuchten Proteine und deren Derivate wurden 2 µl der Probenlösung mit 2 µl der Matrixlösung vermischt und davon 0.5-1.0 µl auf das Target aufgetragen (Dried Droplet). Probenlösungen, die Harnstoff enthielten wurden zweimal mit je 2 µl 0.1% TFA auf dem Target gewaschen.
3.7.6 NMR-Spektroskopie
Die 1H-Kernresonanzspektren wurden an einem 250 MHz-Spektrometer Bruker AC250 aufgenommen. Die Proben wurden in d6-DMSO mit einer Konzentration von etwa 5-10 mg/ml gelöst. Zur Kalibrierung wurde das Lösungsmittel oder Tetramethylsilan (TMS) als interner Standard verwendet. Zur Aufnahme des 1H-NMR-Spektrums von MEL wurden zusätzlich 2-3 Tropfen D2O zur Lösung (NMR-Röhrchen) gegeben.
3.8 Faltungsreaktionen von rhM-CSF
(a) UV-spektroskopische Untersuchung der reduktiven Entfaltung von rhM-CSF
200 µl 0.2 mM MEL (40 nmol) in 10 mM NH4HCO3 pH 8.2 wurden in der UV-Küvette mit 500 µl 6 M Harnstoff, 10 mM NH4HCO3 pH 8.2 und 220 µl 10 mM NH4HCO3 pH 8.2 verdünnt, die Absorptionsmessung gestartet und nach ca. 60 s 163 µg (3.2 nmol) rhM-CSF-Homodimer (2 µg/µl) in 10 mM NH4HCO3 pH 8.2 zugegeben. Nach 75 min wurden 2.4 µl einer 250 mM TCEP-Lösung in H2O zugegeben. Nach 24 h Inkubationszeit wurde ein Aliquot der Reaktionslösung ohne weitere Reinigung, jedoch nach zweimaligem Waschen auf dem MALDI-Target mit 2 µl 0.1% TFA, MALDI-massenspektrometrisch (siehe 3.7.5) charakterisiert.
(b) oxidative Rückfaltungsreaktionen von DTT-reduziertem rhM-CSF
Für die Rückfaltungsreaktion wurde lyophilisiert vorliegendes, dimeres rhM-CSF (400 µg; Fa. Chiron) zunächst mit 260 µl 8 M Harnstoff, 12 mM EDTA, 5 mM DTT, 50 mM Tris HCl pH 8.5 gelöst (cm-csf = 15.4 µg/µl) und für 3 h bei 25 C reduziert.
Diese Lösung wurde in einem Schritt zu 7.5 ml 1 mM EDTA, 0.4 mM GSH, 0.2 mM GSSG 50 mM NH4HCO3 pH 8.5 pipettiert, gemischt und bei 4 C zur in vitro-Rückfaltung inkubiert (cm-csf = 0.5 µg/µl). Unmittelbar nach dem Verdünnen (0 h) und nach 15 min, 1 h, 3 h, 6 h, 18h, 42 h wurden jeweils zwei Aliquote à 200 µg (= 400 µl; 4 nmol) entnommen.
Das jeweils erste Aliquot wurde zu 10 µl einer 1 M IAA-Lösung (10000 nmol) in H2O gegeben und für 1 h bei 25 C im dunkeln inkubiert. Der pH-Wert der Lösung wurde nach 5 und 20 min kontrolliert (mußte aber nicht neu eingestellt werden). Die Alkylierung wurde durch Absenken des pH-Werts auf pH 3 mit 390 µl 0.1% TFA / 0.1% TFA in ACN 6:4 und 10 µl Eisessig (Mischung) abgestoppt (cm-csf = 0.25 µg/µl). In diesem Lösungsmittelgemisch ist rhM-CSF auch im sauren pH-Bereich löslich. 10 µl Reaktionslösung wurden mit 10 µl SDS-Aufschlußpuffer gemischt und bis zur SDS-PAGE (siehe 3.6.4) bei -20 C aufbewahrt. Ein weiters Aliquot der Reaktionslösung wurde MALDI-massenspektrometrisch charakterisiert, der Rest der Lösung sofort tiefgefroren (fl. N2).
Das jeweils zweite 200 µg Aliquot (= 400 µl) der Rückfaltungslösung wurde mit 600 µl einer 0.2 mM MEL-Lösung (120 nmol) umgesetzt (cm-csf = 0.2 µg/µl). MEL war in 5 M Harnstoff, 10 mM NH4HCO3 pH 8.5 gelöst, so daß die Modifizierung in Gegenwart von 3 M Harnstoff erfolgte. Nach ca. 10 min Inkubationszeit bei 25 C wurden 10 µl der Reaktionslösung mit 10 µl SDS-Aufschlußpuffer vermischt bis zur Verwendung bei -20 C (s.o.) aufbewahrt. Ein weiteres Aliquot der Reaktionslösung wurde MALDI-massenspektrometrisch charakterisiert, der Rest der Lösung sofort tiefgefroren (fl. N2). Der molare Überschuß an MEL wurde wie folgt berechnet: MEL reagiert bevorzugt mit Dithiolen zu zyklischen Arsonigsäurediester, d. h. nur das restliche, reduziert vorliegende DTT und die Biscysteinylfunktionen der rhM-CSF Faltungsintermediate bilden stabile MEL-Addukte. Unter Annahme der vollständigen Reduktion von oxidiertem DTT durch GSH betrug das molare Verhältnis von (reduziert vorliegenden) Bisthiolen (DTT und rhM-CSF ) zu MEL 1.05:1.2. Dies entspricht einem molaren Überschuß an MEL von ca. 12%.
Ungefähr 100 µg der carboxamidomethylierten und MEL-modifizierten Faltungsintermediate wurden durch Ultrafiltration (siehe 3.6.2) aufkonzentriert, dreimal mit je 200 µl 0.1% TFA // 0.1% TFA /ACN 6:4 (pH 2) gewaschen und somit in dieses Lösungsmittelgemisch überführt. Dadurch erfolgte neben der Abtrennung aller überschüssigen Reagenzien eine Stabilisierung der MEL-Modifizierung. Das Endvolumen nach Ultrafiltration betrug ca. 80-100 µl (cm-csf 1 µg/µl). Aliquote dieser Lösungen wurden MALDI-massenspektrometrisch charakterisiert.
(c) oxidative Rückfaltungsreaktionen von TCEP-reduziertem rhM-CSF
Für die Rückfaltungsreaktion wurde lyophilisiert vorliegendes, dimeres rhM-CSF (400 µg; Fa. Chiron) zunächst mit 260 µl 8 M Harnstoff, 12 mM EDTA, 5 mM TCEP, 50 mM NaH2PO4 pH 8.5 gelöst (cm-csf = 15.4 µg/µl) und für 3 h bei 25 C reduziert.
Diese Lösung wurde in einem Schritt zu 7.5 ml 1 mM EDTA, 0.4 mM GSH, 0.4 mM GSSG 50 mM NH4HCO3 pH 8.5 pipettiert, gemischt und bei 4 C aufbewahrt (cm-csf = 0.5 µg/µl). Unmittelbar nach dem Verdünnen (0 h) und nach 15 min, 1 h, 3 h, 6 h, 18h, 42 h wurden je zwei 200 µg (4 nmol) Aliquote entnommen.
Die IAA-Alkylierung wurde analog zu (a) durchgeführt. Die Modifizierung mit MEL erfolgte mit 600 µl 0.1 mM MEL-Lösung (60 nmol) für 10 min bei 25 C. MEL war in 5 M Harnstoff, 10 mM NH4HCO3 pH 8.5 gelöst, so daß die Modifizierung in Gegenwart von 3 M Harnstoff erfolgte. Alle weiteren MS-Analysen und Reinigungsschritte wurden analog (a) durchgeführt.
3.9 Computerprogramme
Die Berechnungen der Molekulargewichte der nach proteolytischen Abbaureaktionen zu erwartenden Fragmente erfolgte mit dem Programm GPMAW 2.1.0.
Zur Berechnung der Solvent Accessible Surface innerhalb des Programm INSIGHT II (Fa. Biosym) wurde ein Makro geschrieben, daß ein komfortables Erstellen der Eingabeparameter (Anzahl und Art der zu berechnenden Atome) erlaubt und die berechneten Ergebnisse zusammenfaßt [160].
|
|
|
|
|
|
|
|
2.8 Computerberechnungen von lösungsmittelzugänglichen Oberflächen in Proteinen mit bekannter Struktur |