|
|
|
|
|
1.3 Massenspektrometrische Methodenzur Charakterisierung von proteinchemischen Umsetzungen |
|
|
1.1 Cysteinylthiolgruppen und Disulfidstrukturen in Proteinen |
1.2 Derivate der Arsonigen Säure als chemoselektive Modifizierungsreagenzien
Die Toxizität und z.T. die pharmakologische Wirkung von Arsenverbindungen ist schon seit Jahrtausenden bekannt. So empfahl bereits Hippocrates (460-377 v. Chr.) einen Brei aus Realgar (As2S4) zur Behandlung von Geschwüren. Im 15 bis zum 17 Jahrhundert verwendeten v.a. die Adelshäuser in Europa Arsen(III)oxid (Arsenik) zum Entledigen unbeliebter Konkurrenten [46]. Die erste synthetisierte Organoarsenverbindung Kakodyl wurde 1760 von L. C. Cadet synthetisiert. Jedoch gelang es erst Bunsen (1837-1842) die Reaktion von Cadet aufzuklären. Damit war der Grundstein für allgemeine Synthesen von Organoarsenverbindungen gelegt. Bahnbrechende Arbeiten von Paul Ehrlich (1905) führten zur sogenannten "Reduktionstheorie", nach der fünfwertige Arsenverbindungen in vivo zuerst reduziert werden müssen, d.h. die eigentlich giftigen bzw. therapeutisch wirksamen Substanzen sind die Arsen(III)verbindungen. Paul Ehrlich vermutete, daß die therapeutisch eingesetzten Arsenverbindungen an definierten Strukturen - den sog. "Arsenoceptoren" - des Krankheitserreger binden müssen, um ihre Wirkung zu entfalten [47]. Mit der Synthese des Salvarsans war es erstmals möglich den Erreger der Syphilis direkt zu bekämpfen. Die Entwicklung dieses Medikaments gilt allgemein als der Beginn der modernen Chemotherapie [48]. Bis zum Jahr 1932 wurden ca. 12500 Organoarsenverbindungen synthetisiert und auf ihre biologische Aktivität überprüft, u.a. Arsphenamin (1910) und Oxophenarsin (1932). Mit der Entdeckung des Penizillins verloren die Organoarsenverbindungen aufgrund ihrer hohen Toxizität rasch an Bedeutung. Nur Melarsoprol wird heute noch in schweren Fällen der Schlafkrankheit therapeutisch eingesetzt (siehe S. 11). Während des ersten Weltkrieges fand sich für die Organoarsenverbindungen ein weniger ruhmreiches Anwendungsgebiet, das der chemischen Kampfstoffe, von denen Lewisite I (2-Chlorvinylarsindichlorid) am bekanntesten ist. Da insbesondere Dithiole als Antidot gegen Arsenvergiftungen wirkten, war der Rückschluß nahe, daß die Toxizität trivalenter Organoarsen(III)verbindungen auf der Wechselwirkung mit Proteinthiolen beruht. Die Beschreibung dieser Wechselwirkungen erfolgt ausführlicher in Abschnitt 1.2.3. Zuvor soll näher auf die Nomenklatur (siehe 1.2.1) und auf diechemischen, strukturellen Eigenschaften(siehe 1.2.2)) der in dieser Arbeit verwendeten Organoarsenverbindungen (siehe 2.1) eingegangen werden.
1.2.1 Nomenklatur von trivalenten Organoarsen(III)verbindungen
Die uneinheitliche Benennung organischer Arsenverbindungen beruht zum einen auf einer historischen Analogie zu den Stickstoff-Verbindungen und zum anderen auf der Empfehlung von IUPAC und Chemical Society die Benennung der Arsenderivate in Analogie zu den Organophosphor-Verbindungen durchzuführen. Letztere Regeln lassen sich nicht ohne weiteres aus dem englischen in den deutschen Sprachgebrauch übertragen; somit existieren fast immer zwei gleichwertige Namen [48]. Auch erfolgt in der Literatur die Zuordnung von Oxidationszahlen nicht konsequent [46]. Für die Elektronegativitäten EN(C) = 2.5, EN(H) = 2.2 und EN(As) = 2.0 [49] ergibt sich folgende Einteilung: Arsen in trivalenten Organoarsenverbindungen besitzt die Oxidationszahl +3, solange keine As-H Bindungen enthalten sind; gleiches gilt für pentavalente Verbindungen mit der Oxidationsstufe +5. Dagegen wird dem Arsen in AsH3 (1) die Oxidationszahl -3 zugewiesen.
Analog den Aminen und Phosphinen werden von As-IIIH3 (1) abgeleitete Verbindungen als Arsinebezeichnet (siehe Abb. 2). Davon abgeleitete Verbindungen, die weitere Heteroatome (z.B. Halogene) enthalten, lassen sich als Abkömmlinge der Arsine (1) oder als Derivate der Arsonigen Säure (3) (arsonous acid) auffassen. Obwohl die freie Arsonige Säure nur schwache Säureeigenschaften zeigt, verhalten sich Organo-arsonigsäure-dichloride (2) gegenüber Alkoholen oder Aminen wie Säurechloride.

Abb. 2: Strukturformeln einiger Arsen(III)-Verbindungsklassen.
Die Bezeichnung Dihydroxy-organo-arsin für3 ist dennoch gerechtfertigt, da das Tautomergleichgewicht zwischen der Arsonigen Säure (3) (arsonous acid) und der Arsinsäure (4) (arsinic acid) vollständig auf der Seite der Dihydroxy-Form 3 liegt (siehe Abb. 3). Ähnliches gilt für die Arsenige Säure (5) (arsenious acid) und die Arsonsäure (6) (arsonic acid).

Abb. 3: Arsen-Sauerstoffsäuren und tautomere Formen. Das Tautomergleichgewicht liegt jeweils vollständig auf der Seite der Hydroxyformen 3 und5.
Wie weiter oben schon erwähnt ändern sich die formalen Oxidatiosstufen bei der Einführung direkt an das Arsen gebundener organischer Reste. In Tab. 2 sind die unterschiedliche Benennung einiger Derivate gegenübergestellt.
Tab. 2: Nomenklatur einiger Derivate, die sich von den Arsonigen Säuren bzw. Arsinen ableiten.
|
Derivat |
Name als Säure-Derivat |
Name als Arsin-Derivat |
angelsächsisch |
|
R-As (OR)2 (7) |
Organo-arsonigsäure-diester |
Dialkoxy-organo-arsin |
Arsonous acid diester |
|
R-As (NH2)2 (8) |
Organo-arsonigsäure-diamid |
Diamino-organo-arsin |
Arsonous diamide |
|
R-As (SR)2 (9) |
Organo-arsonigsäure- |
Dimercapto-organo-arsin |
Thioarsinite, |
1.2.2 Chemische, physikalische und strukturelle Eigenschaften der Arsonigen Säure und ihrer Anhydride
Eine in der Literatur vielfach beschriebene Stoffklasse sind die Anhydride der arsonigen Säurederivate. Das ursprünglich angenommene Strukturelement R-As=O (10) führte in Analogie zur Nitroso-Gruppe zur Bezeichnung Arsenoso-organyl. Auch die Bezeichnungen Organo-arsen-oxid (arsine oxide) und Oxo-organo-arsin(oxo-organo-arsine) werden verwendet. Eine Vielzahl der Anfang dieses Jahrhunderts hergestellten Verbindungen wurden v.a. auf ihren therapeutischen Nutzen überprüft, sind aber weder ausreichend charakterisiert noch in ihrer Struktur gesichert. Die Formulierung R-As=O ist insoweit inkorrekt, als diese Verbindungen weitgehend oligomer vorliegen (sieheAbb. 4). Die Bezeichnung Arsenoso - in Analogie zu Nitroso - ist nicht geeignet, da keine chemischen und strukturellen Analogien zur Nitroso-Gruppe bestehen. Auch die Bezeichnung Organo-arsen-oxid ist irreführend, da keine As=O Doppelbindung vorliegt. Sowohl von ihrer Herstellungsmethode als auch ihrem chemischen Verhalten entsprechen diese Verbindungen eher Abkömmlingen der Arsonigen Säuren (3) bzw. ihrer oligomeren Säureanhydride [RAsO]n (11). [48]
Arsonige Säuren (3) stehen in wäßrigen Systemen im Gleichgewicht mit ihren Anhydriden (11). Die Lage des Gleichgewichts wird hauptsächlich durch den organischen Rest R bestimmt (sieheAbb. 4). Für R = Alkyl liegt ausschließlich das oligomere Anhydrid vor. Für R = Aryl beeinflussen evt. vorhandene Substituenten am Aromaten, sowie die Isolationsbedingungen das Gleichgewicht [51].
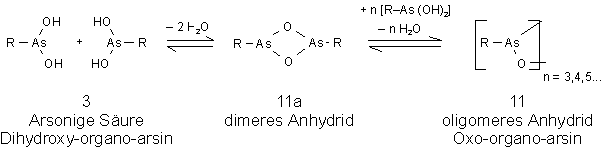
Abb. 4: Gleichgewicht der freien Arsonigen Säure (3) mit ihren (oligomeren) Anhydriden (11).
Phenylarsonige Säure (12) bzw. ihr Anhydrid [PhAsO]n (12a) (phenylarsonicacid, PAA; auch Phenylarsenoxid, PAO vgl. S. 4) liegt in Form oligomerer Anhydride mit n = 2-5 vor. In Benzol und Campher ebullioskopisch und kryoskopisch bestimmte Molekulargewichte bestätigen ein Tetramer [52]. Das in Abb. 5gezeigte Elektronenstoß(EI)-Massenspektrum von PAA zeigt zyklische Oligomere für n = 3-5. IR-spektroskopischen Untersuchungen zeigen für die As-O Einfachbindung eine intensive und breite Absorption im Bereich ![]() = 715-750 cm-1; damit bestätigt sich die As-O-As Ringstruktur [53]. In Analogie dazu kristallisiert [PhAsS]n als zyklisches Tetramer mit As-S-As Einfachbindungen [54] (Bindungslängen siehe Tab. 3). Aus Gründen der Übersichtlichkeit wird im weiteren nur noch der Begriff Arsonige Säure verwendet, soweit nicht eine strukturelle Unterscheidung von freier Arsoniger Säure und ihrem Anhydrid notwendig erscheint.
= 715-750 cm-1; damit bestätigt sich die As-O-As Ringstruktur [53]. In Analogie dazu kristallisiert [PhAsS]n als zyklisches Tetramer mit As-S-As Einfachbindungen [54] (Bindungslängen siehe Tab. 3). Aus Gründen der Übersichtlichkeit wird im weiteren nur noch der Begriff Arsonige Säure verwendet, soweit nicht eine strukturelle Unterscheidung von freier Arsoniger Säure und ihrem Anhydrid notwendig erscheint.
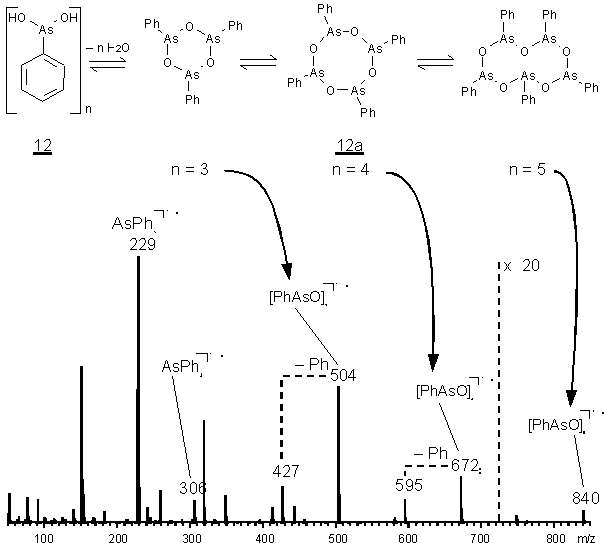
Abb. 5: EI-MS der oligomeren zyklischen Anhydride (12a) der Phenylarsonigen Säure (12) (PAA) sowie ihrer Fragmentionen.
Im folgenden soll kurz auf die chemische Reaktivität der Arsonigen Säuren (3) eingegangen werden, soweit diese mit Modifizierungen von Proteinen und deren Charakterisierung im Zusammenhang steht. Im Hinblick auf die Charakterisierung von Faltungsintermediaten spielt das Redoxverhalten der in der Oxidationszahl +3 vorliegenden Arsonigen Säuren (3) eine Rolle. Nur in stark alkalischer Lösung (1 N NaOH) reduzieren Derivate der Phenylarsonigen Säure (12) Cystine zu Cysteinen, wobei sie selbst zu Arsonsäuren-Derivate (6) oxidiert werden [55]. Die Reaktion der Arsonigen Säuren (3) mit Thiolen wird separat in Abschnitt1.2.3 behandelt. Als mögliche Nebenreaktion der Modifizierung von Proteinen mit Arsonigen Säuren (3) kommen O- und N-Modifizierungen der Aminosäuren Serin, Threonin, Tyrosin, Lysin, Arginin und Histidin in Betracht. Die direkte Umsetzung zu offenkettigen Organo-Arsonigsäurediester (7) gelingt durch Erhitzen des Anhydrids (11) mit Alkoholen unter CaCl2 [56]. Auch zyklische Organo-Arsonigsäurediester (7a) mit Diolen, z.B. Glykol sind bekannt [57]. Die resultierenden Organo-arsonigsäure-diester (7) hydrolysieren und oxidieren bereits durch Luftsauerstoff zu Ester der Arsenigen Säure (5) [51]. Dichlororganoarsine (2) reagieren unter Ausschluß von Wasser bereitwillig mit Ammoniak und Aminen zu Organoarsonigsäurediamiden (8). Die As-N bzw. As=N Bindung hydrolysiert jedoch spontan mit Wasser; auch mit Alkoholen oder Thiolen erfolgt die Spaltung zu den entsprechenden Arsonigsäurediester (7) bzw. -dithioester (9) [51]. Analog hydrolysieren gemischte O-/S-Diester in wäßrigen Systemen zur Arsonigen Säure und dem entsprechenden Thiol [48]. Keine der hier genannten Reaktionen kommt daher als störende Nebenreaktion bei den proteinchemischen Umsetzungen in nennenswertem Ausmaß (siehe 2.2) in Betracht.
1.2.3 Chemische Reaktionen Arsoniger Säuren mit Dithiolen
Erst 1923 zeigte Vögtlin et al., daß die Toxizität von Arsen(III)verbindungen auf der Wechselwirkung des Arsens mit Thiolkomponenten lebender Zellen beruhte. Mit der Erkenntnis, daß Thiole als Antagonisten der Arsentoxizität wirkten, wurde die Toxizität auf die Inhibition des Glutathionstoffwechsels und/oder thiolhaltiger Enzyme ("Arsenoceptoren" (siehe S. 5)) zurückgeführt [58]. Obwohl dies heute weitgehend akzeptiert ist, besteht weiterhin Unklarheit über den exakten Wirkmechanismus. So wird zwar die Inhibition vieler Enzyme durch trivalente Organoarsenverbindungen der Wechselwirkung mit - im aktiven Zentrum befindlichen - Cysteinylthiolgruppen zugeordnet (s.u.), jedoch existieren keinerlei Kristallstrukturen von Peptiden oder Proteinen die diese Wechselwirkung strukturell charakterisieren. Demgegenüber sind Wechselwirkungen mit niedermolekularen (Di-)Thiolen genauer charakterisiert.
Mono- und Dithiole reagieren mit Arsonigen Säuren (3) (auch in Form ihrer Anhydride (11)) zu offenkettigen (9a) oder zyklischen (9b) Arsonigsäuredithioester (sieheAbb. 6). Der exakte Reaktionsmechanismus ist unbekannt. Vermutlich ist die freie Arsonige Säure die aktive Form, da Dichlororganoarsine (2) schnell mit Thiolen zu 9a oder 9b reagieren.
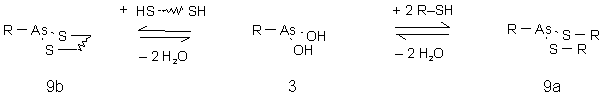
Abb. 6: Allgemeines Reaktionsschema für die Reaktion Arsoniger Säuren (3) mit Mono- und Dithiolen zu offenkettigen (9a) und zyklischen (9b) Arsonigsäuredithioestern.
Untersuchungen von Cohen et al. [55] an Derivaten der Phenylarsonigen Säure mit Monothiolen (Cysteine, Glutathion u.a.) zeigen, daß die Thioester (9a) in saurer und schwach alkalischer Lösung (NaHCO3) stabil sind, aber schnell in Gegenwart von 0.1 N NaOH zur entsprechenden Arsonigen Säure (3) hydrolysieren. Sie schlagen ein reversibles Hydrolysegleichgewicht vor (siehe Abb. 7).
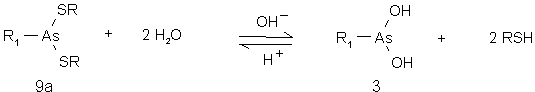
Abb. 7: Allgemeines Reaktionsschema für die basische Hydrolyse von Arsonigsäuredithioestern (9a) zur Arsonigen Säure (3) und dem entsprechenden Thiol.
Aus Untersuchungen über Thiole als Antidot gegen Arsenvergiftungen geht hervor, daß die zyklischen Arsonigsäuredithioester (9b) deutlich stabiler sind, als die Addukte aus monosubstituierten Arsen(III)verbindungen [58]. V. P. Whittaker zeigte 1947, daß die Ausbildung des Fünfringes - resultierend aus der Reaktion mit 1,2-Dithiolen - geringfügig dem Sechsring bevorzugt ist [59]. Neuere NMR-spektroskopische Untersuchungen bestätigen dies. [60]
Aufgrund der niedrigen Inversionsrate des pyramidal gebauten Arsenatoms erhält man für die Reaktion unsymmetrisch substituierter Dithiole - also immer auch für Biscysteinylgruppierungen in Proteinen - mit Arsonigen Säuren (3) diastereomere zyklische Arsonigsäuredithioester (9b). Man kann syn und anti im Bezug zum organischen Rest am Arsen und am Dithiol unterscheiden. Die Diastereomere sind durch HPLC-Trennung isolierbar (siehe 2.3.2). InAbb. 8 sind die Diastereomere des Addukts (13) aus Tolyldichloroarsin (TDA) und 1,2-Dimercaptopropanol (DMP; auch British Anti Lewisite, BAL) gezeigt. Sowohl NMR-spektroskopische Untersuchungen [61], als auch die Röntgenstruktur des anti-Isomers [62] belegen die thermodynamische Bevorzugung des Anti-Addukts aufgrund sterischer Wechselwirkungen. Unter sauren Bedingungen beobachtet man eine rasche Gleichgewichtseinstellung beider Isomere, wobei weniger die Inversion am Arsenatom eine Rolle spielt. Vielmehr wird eine Ringöffnung (As-S Bindung) und anschließende Rückbildung der Ringstruktur vermutet [60].
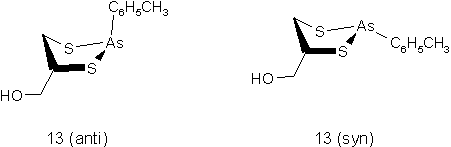
Abb. 8: Diastereomere des TDA-DMP-Addukts (13). Syn und Anti beziehen sich auf die organischen Reste der Arsonigen Säure und des Dithiols.
In Tab. 3 sind As-S Bindungslängen und S-As-S Bindungswinkel bekannter Röntgenstrukturen zusammengefaßt.Abb. 9 zeigt schematisch den durchschnittlichen Abstand von 3.49 Å zweier durch Arsen verbrückter Schwefelatome (berechnet von den durchschnittlichen Bindungsabständen und -winkel aus Tab. 3). S-S-Bindungsabstände in Disulfidbrücken von Proteinen liegen im Bereich kovalenter S-S-Einfachbindungen: 2.05-2.15 Å [49].
Tab. 3: As-S Bindungslängen und S-As-S Bindungswinkel von strukturell charakterisierten Organoarsen-Schwefel-Verbindungen.
|
Verbindung |
As-S [Å] |
S-As-S [] |
Referenz |
|
"Kakodyl Disulfid" (Me2As(=S)SAsMe2) |
2.214 / 2.075 2.075 (As=S) |
113.3 |
[63] |
|
Diphenyldiarsendisulfid (Ph2As2S3) |
2.252 / 2.253 |
98.4 |
[64] |
|
5-Chlor-1-oxa-4,6-dithia-5-arsocan |
2.249 |
104.0 |
[65] |
|
Cyclo-tetrakis(phenylarsensulfid (PhAsS)4 |
2.262 / 2.25 |
102.1 |
[54] |
|
1,3-Dithia-2-phenylarsino-[3]ferrocenophan |
2.252 / 2.255 |
98.6 |
[66] |
|
Tolylarsen-2,3-dimercaptopropanolat |
2.225 / 2.276 |
92.7 |
[62] |
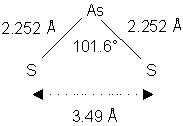
Abb. 9: Geometrisch berechneter Abstand zweier über Arsen verbrückter Schwefelatome.
Die erstmals 1942 von E. Friedheim [67] synthetisierte Verbindung p-(4,6-Diamino- s-triazin-2-yl) aminophenylarsonige Säure (14) - mit dem Trivialnamen Melarsen Oxid (MEL) (Strukturformel siehe 2.1) - wird noch heute als Medikament in Form des Addukts mit 1,2-Dimercaptopropanol (Melarsoprol) zur Behandlung später Stadien der afrikanischen Schlafkrankheit (griech. Trypanosomiasis) eingesetzt. Dies ist eine in weiten Teilen des tropischen Afrikas herrschende Infektionskrankheit deren Erreger Trypanosomen sind. Diese parasitären Geißeltierchen leben im Blut, Lymphsystem und in der Gehirn-Rückenmarksflüssigkeit von Säugetieren. Die Übertragung der Krankheit erfolgt durch die Tsetsefliege. Im Endstadium der Krankheit kommt es zu nervlichen und seelischen Schädigungen, schließlich zurSchlafsucht Die Trypanosomen reagieren äußerst empfindlich auf trivalente aromatische Organoarsenverbindungen, da sie über ein spezifisches in Säugerzellen nicht vorkommendes Transportsystem aufgenommen werden [68]. Insbesondere Melarsen Oxid führt zur schnellen Lyse der Trypanosomen [69]. Der Wirkmechanismus dieser Verbindung ist Gegenstand aktueller Forschung [70, 71]. Vermutlich inhibiert Melarsen Oxid das System von Trypanothion (N1,N8-Bis(glutathionyl)spermidin) - einem intrazellulären Dithiol - und der Trypanothion-Reduktase, das ähnlich Funktionen erfüllt wie das System Glutathion / Glutathion Reduktase anderer Organismen [72]. Das von A. H. Fairlamb et al. 1989 durch HPLC isolierte Addukt aus Melarsen undTrypanothion stellt den wohl ersten makrozyklischen Arsonigsäuredithioester dar, der massenspektrometrisch (PD-MS) charakterisiert wurde [69].
Proteine , von denen bekannt ist, daß sie benachbarte Thiolgruppen enthalten werden durch Arsonige Säuren (3) effizient inhibiert. Analog niedermolekularen Dithiolen begründet man dies mit der Ausbildung zyklischer Arsonigsäuredithioester (9b) (sog. Ringhypothese der Arsentoxizität [59], sieheAbb. 6 bzw. Abb. 10). Die Inhibition ist reversibel und kann durch Dithiole, wie 1,2-Dimercaptopropanol (DMP) oder 1,4-Dithiothreitol (DTT) (sieheAbb. 35) aufgehoben werden. Monothiole sind dafür deutlich schlechter geeignet, da sie nur offenkettige Arsonigsäuredithioester (9a) bilden können [59]. Umgekehrt wird aus dem analogen Verhalten unbekannter Proteine auf die Anwesenheit räumlich naher Cysteinreste geschlossen.
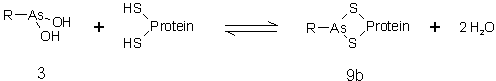
Abb. 10: Bildung zyklischer Arsonigsäuredithoester (9b) aus Proteinthiolen und Arsoniger Säure (3).
Typische Inhibitoren im µM-Konzentrationsbereich sind Phenylarsonige Säure (12) [73], 4-Aminophenylarsonige Säure (15) [74] und 4-(Bromoacetyl)aminophenylarsonige Säure [75]. Die quantitativen Bestimmung der Arsenmodifizierung erfolgt überwiegend mit radioaktiv markierten Derivaten, z.B. p-125Iodphenylarsonige Säure [76]. Nur von wenigen Enzymen(komplexen) glaubt man die für die Wechselwirkung mit trivalenten Organoarsen(III)verbindungen verantwortlichen Thiolkomponenten identifiziert zu haben. Dazu gehört das vicinale Dithiol Liponsäure im Pyruvat-Dehydrogenase-Komplex [74], die Cysteine 31 und 184 der Lecithin-Cholesterol-Acyl-Transferase [75] und die zwei räumlich benachbarten Cysteinen im aktiven Zentrum von Thioredoxin [77]. Für die meisten Enzyme, die eine Reaktion mit Arsonigen Säuren (3) eingehen, steht die exakte strukturelle Charakterisierung der Arsen(III)bindungsstelle noch aus [58].
|
|
|
|
|
1.3 Massenspektrometrische Methodenzur Charakterisierung von proteinchemischen Umsetzungen |
|
|
1.1 Cysteinylthiolgruppen und Disulfidstrukturen in Proteinen |
Diese Seite wurde erstellt am 15. März 1998. Letzte Änderungen am 30. Juni 1998 durchPeter Happersberger.