|
|
|
|
|
2.2 UV-spektroskopische Untersuchungen zum Reaktionsmechanismus der Modifizierung von Bis-Thiolen mit Melarsen Oxid |
|
|
1.5 Zielsetzung und Aufgabenstellungen der Arbeit |
2. Allgemeiner Teil
2.1 Synthese von Derivaten der Arsonigen Säure
Es sollten Derivate der Phenylarsonigen Säure (12) (PAA) hergestellt werden, die zum einen deutlich besser als PAA (12) in Wasser oder Wasser/Methanol-Gemischen löslich sind, und zum anderen es ermöglichen, die Modifizierungsreaktion UV-spektroskopisch zu verfolgen. Folgende Derivate wurden ausgewählt: das Anhydrid der Pyridin-3-arsonigen Säure (16) (PYR) und die p-(4,6-Diamino-s-triazin-2-yl)aminophenylarsonige Säure (14) (MEL; Melarsen Oxid). Die Verbindung 4-Aminophenylarsonige Säure (15) (A-PAA) wurde von Tilman Schmachtel [119] zur Verfügung gestellt. In Abb. 18 sind die Strukturformeln zusammengefaßt.
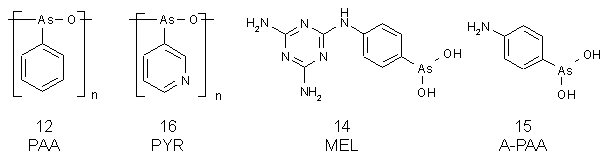
Abb. 18: Strukturformeln der in dieser Arbeit untersuchten Derivate der Arsonigen Säure.
PYR (16) entspricht sterisch nahezu PAA (12). Es sollte durch die Einführung eines Wasserstoffbrücken-Akzeptors (Ringstickstoff) deutlich besser löslich als PAA (12) sein. Der aromatische Ring ist elektronenärmer und der zur Arsenfunktion meta stehende Ringstickstoff kann mit der Arsenfunktion keine mesomere Wechselwirkung eingehen.
Die Aufnahme von MEL (14) in die Auswahl beruhte vor allem aus bisherigen Untersuchungen von A. H. Fairlamb [69], der das Addukt aus MEL (14) und Trypanothion isolieren und charakterisieren konnte (siehe 1.2.3). MEL (14) ist sterisch deutlich anspruchsvoller als PAA (12). Der aromatische Ring der Arsenfunktion ist durch die (substituierte) Aminogruppe bei MEL (14) und A-PAA (15) elektronenreicher als bei PAA (12). Die deutlich höhere Löslichkeit von A-PAA (14) gegenüber PAA (12) in organischen Lösungsmitteln war bereits bekannt [119].
2.1.1 Synthese des Anhydrids von Pyridin-3-arsoniger Säure (PYR)
Pyridin-3-arsonigsäuredichlorid Monochlorhydrat
In der von A. Binz und O. v. Schickh [120] 1936 beschriebenen Synthese wird von 3-Aminopyridin (17) ausgegangen, welches mit Arsen(III)chlorid (18) und Natriumnitrit in Gegenwart von Kupfer(I)chlorid (Katalysator) zu Pyridin-3-arsintetrachlorid (19) umgesetzt wird. Die Reduktion zum Pyridin-3-arsonigsäuredichlorid Monochlorhydrat (20) erfolgt nach Abtrennung von Natriumchlorid und Kupferchlorid mit Schwefeldioxid und Kaliumiodid als Katalysator (siehe Abb. 19). Das eigentlich reduzierend wirkende Reagenz ist das Iodid-Anion, das zu Iod oxidiert wird. Schwefeldioxid reduziert dann Iod wieder zu Iodid.
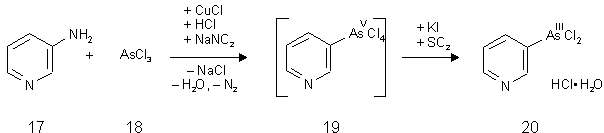
Abb.19:Synthese von Pyridin-3-arsonigsäuredichlorid Monochlorhydrat (20)[120]
Diese sog. Scheller-Reaktion (1931) ist eine Modifizierung der Bart-Reaktion, die ähnlich der Sandmeyer-Reaktion (Halogenierung von Aromaten) verläuft [121]. Das als Niederschlag ausfallende Pyridin-3-arsonigsäuredichlorid Monochlorhydrat (20) wurde abgetrennt und nach mehrmaligem Umkristallisieren und Sublimation im Feinvakuum als feinkristalliner, farbloser Feststoff erhalten. Die Dünnschichtchromatographie zeigte ein reines Produkt.
Das EI-Massenspektrum von Pyridin-3-arsonigsäuredichlorid (20) (siehe Abb. 21) zeigt das monomere Molekularion (m/z 223) mit einer für zwei Chloratome typischer Isotopenverteilung von 10:6.4:1 (für m/z 223, 225, 227) [122], zusammen mit den Fragmentionen. InAbb. 20 ist das Fragmentierungsschema dargestellt. Der Basispeak - das Pyridinyl-Kation (m/z 78) - entsteht aus der Abspaltung von AsCl2 (m/z 145). Die aus einer der Benzylspaltung analogen Fragmentierung entstehenden Azarsepinen (m/z 153 und 188) spalten Blausäure (HCN) und Acetylen ab. Man erhält so Arsole (m/z 126 und 161) und Arsirene (m/z 100 und 135).
Das 1H-NMR Spektrum von Pyridin-3-arsonigsäuredichlorid Monochlorhydrat (20) ist in Abb. 23 A dargestellt. Für Verbindung20 lassen sich die Signale eindeutig zuordnen: ein Triplett bei 8.08 ppm (H3), zwei Dupletts bei 8.82 ppm (H4) und 9.09 ppm (H2) und ein Singulett bei 9.20 ppm (H1). Das Spektrum wurde in d6-DMSO aufgenommen und internmit TMS kalibriert.
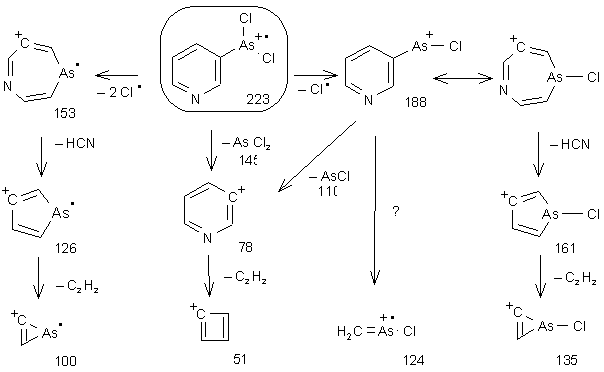
Abb. 20: EI-MS Fragmentierungsschema und m/z Werte der Fragmentionen von Pyridin-3-arsonigsäuredichlorid (20).

Abb. 21: EI-MS Spektrum von Pyridin-3-arsonigsäuredichlorid (20).Die vorgeschlagenen Strukturen der Fragmentionen sind Abb. 20 dargestellt.
Anhydrid der Pyridin-3-arsonigen Säure
Das erhaltene Pyridin-3-arsonigsäuredichlorid Monochlorhydrat (20) wird durch Zugabe von 25%igem Ammoniak zunächst zur freien Arsonigen Säure (21) hydrolysiert (sieheAbb. 22).
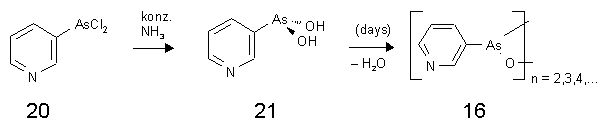
Abb. 22:Synthese des Anhydrids der Pyridin-3-arsonigen Säure (16) (PYR)[120].
Nach längerem Stehenlassen scheidet sich das Anhydrid Pyridin-3-arsonigen Säure (16) als feinkristalliner, farbloser Feststoff ab, der chromatographisch einheitlich war. Die CHN-Elementaranalyse stimmte - unter Einbezug eines Moleküls Kristallwassers - mit den theoretisch berechneten Werten überein.
Der aromatische Bereich des 1H-NMR Spektrums von 16 ist inAbb. 23 B dargestellt. Zum Vergleich sind die chemischen Verschiebungen für Pyridin (22) und 3-Aminopyridin (17) angegeben. Man erhält - im Vergleich zu 22 - ein deutlich komplizierteres Spektrum.

Abb. 23: Das1H-NMR Spektren von (A) Pyridin-3-arsonigsäuredichlorid Monochlorhydrat (20) und B) vom Anhydrid bzw. der freien Arsonigen Säure (16) (PYR); beide in d6-DMSO aufgenommen. Die interne Kalibrierung erfolgte durch TMS. Die chemischen Verschiebungen für Pyridin (22) und 3-Aminopyridin (17) sind zusätzlich angegeben.
Die gleich großen Integrale der zu Multipletts aufgespaltenen Signale (7.47, 8.14, 8.63 und 8.75 ppm) lassen auf ein überlagertes Spektrum unterschiedlicher oligomerer Anhydride (16) schließen. Das Singulett bei 6.97 ppm könnte den OH-Protonen von z. T. in freier Form vorliegender Pyridin-3-arsoniger Säure (21) entsprechen. Alle Signale sind im Vergleich zu17 und sogar zum Pyridin (22) tieffeldverschoben, d.h. die funktionelle Gruppe Arsonige Säure besitzt einen -M (und/oder -I) Effekt.
Das EI-Massenspektrum (siehe Abb. 24) von PYR (16) zeigt als Molekularion und Basispeak (m/z 507) das trimere, zyklische Arsonigsäureanhydrid. Geringere Intensitäten zeigen das tetramere (m/z 676), dimere (m/z 338) und monomere (m/z 169) Anhydrid. Es existieren keine Signale für die freie Arsonigen Säure (m/z 187). Es treten Fragmentierungen durch Abspaltung eines Pyridinylrestes (m/z 78) für das Tetramer (m/z 598), Trimer (m/z 429) und Dimers (m/z 260) auf. Weitere Fragmente sind AsO+ (m/z 90.9), [AsO]2+ (m/z 181.8) und [(C5H4N)2As]+ (m/z 231).
Das ESI-Massenspektrum (siehe Abb. 26) von16 bestätigt nochmals den oligomeren Charakter dieser Verbindung im wäßrigen System 2 % AcOH / MeOH 9:1. Man erkennt eine Oligomerverteilung (PYR)n für n = 2-11 mit einem Intensitätsmaximum für das Tetramere. Der sterisch gespannte Vierring des zyklischen Dimers (m/z 339) ist weniger bevorzugt als die offenkettige Form (m/z 357). Ab dem Trimer ist die cyclische (m/z 508) der offenkettigen Form (m/z 526) bevorzugt. Die Massendifferenz von m = 18 zwischen der zyklischen und offenkettigen Form zeigt eindeutig, daß keine Methylester (m = 14) vorliegen. In Abb. 25 sind diese Gleichgewichte schematisch dargestellt.
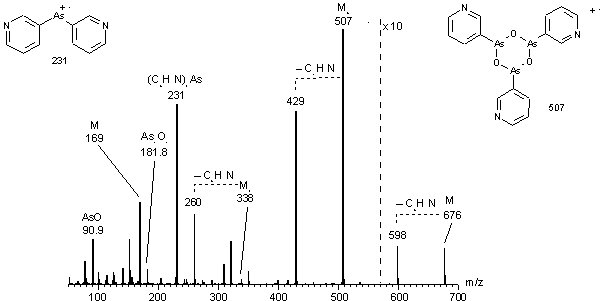
Abb. 24: EI-MS Spektrum vom Anhydrid der Pyridin-3-arsonigen Säure (PYR) (16). Deutlich erkennt man oligomere Molekülionen M3+ und M4+. Für das trimere Anhydrid (m/z 507) und das Fragmention m/z 231 ist die Strukturformel angegeben.
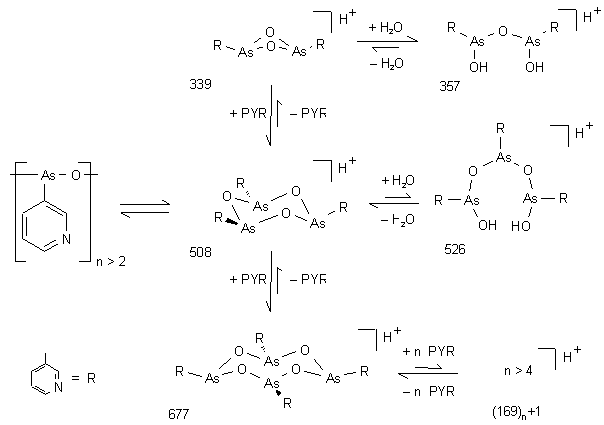
Abb. 25: Vorgeschlagene Gleichgewichtsstrukturen des (oligomeren) Anhydrids der Pyridin-3-arsonigen Säure (16) (PYR) unter ESI-MS Bedingungen. Die angegebenen m/z Werte entsprechen den protonierten Molekülionen.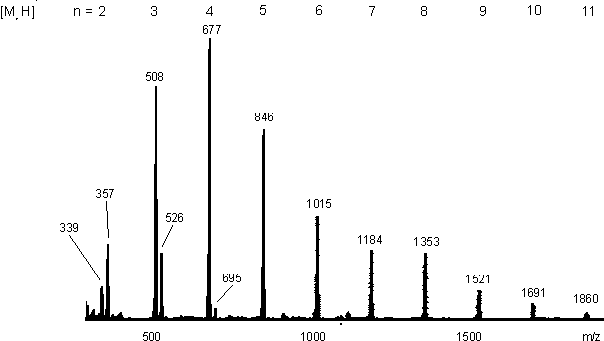
Abb. 26: ESI-MS Spektrum vom Anhydrid der Pyridin-3-arsonigen Säure (16) (PYR) in 2% AcOH/MeOH 9:1 (cpyr = 30 mM). Die Serie zyklischer oligomerer Anhydride [MnH]+ reicht von n = 2-11. Außerdem sind offenkettige oligomere Anhydride für n = 2-6 zu erkennen. Die entsprechenden Gleichgewichtsstrukturen zeigt Abb. 25.
Das FAB-Massenspektrum (Matrix: Glycerin / DMSO / AcOH 1:1:1) bestätigt den chemischen Charakter der Arsonigen Säure. Unter Bedingungen der FAB-MS-Analyse (saures Mileau, großer Überschuß an Glycerin, Hochvakuum) reagiert PYR (16) mit Glycerin zu offenkettigen oder zyklischen Diestern. In Abb. 29 sind die analogen Strukturen für MEL gezeigt. Es zeigen sich Signale für das monomere Anhydrid der Pyridin-3-arsonigen Säure (16), oligomere Anhydride mit n > 1 liegen nicht vor. Bei Verwendung einer Matrix mit einem monofunktionellen Alkohol (3-Nitrobenzylalkohol (NBOH) / DMSO / AcOH 1:1:1) erhält man als Basispeak den offenkettigen Pyridin-3-arsonigsäure-bis-(3-nitrobenzyl)ester und keine höhermolekularen Addukte. Entsprechend zeigt das FAB-MS Spektrum für die alkoholfreie Matrix 2-Nitrophenyl-n-octylether / DMSO / AcOH 1:1:1 nur das tetramere, protonierte Anhydrid (16). Offensichtlich werden die oligomeren Anhydride (18) durch die Reaktion mit Alkoholen in ihre monomolekularen Diester überführt. Gleiches ist für die Reaktion mit Thiolen zu erwarten.
2.1.2 Synthese von p-(4,6-Diamino-s-triazin-2-yl)aminophenylarsoniger Säure (Melarsen Oxid, MEL)
p-(4,6-Diamino-s-triazin-2-yl)aminophenylarsonsäure (Melarsen)
Die Umsetzung von 2-Chlor-4,6-diamino-1,3,5-triazin (23) mit p-Aminophenylarsonsäure (24) erfolgt unter leicht sauren Bedingungen zur p-(4,6-Diamino-s-triazin-2-yl)aminophenylarsonsäure (Melarsen) (25) (siehe Abb. 27) [124, 125].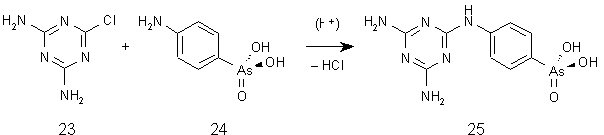
Abb. 27: Darstellung von p-(4,6-Diamino-s-triazin-2-yl)aminophenylarsonsäure (Melarsen) (25).
Man erhält einen farblosen, amorphen Feststoff, der in Wasser und organischen Lösungsmitteln schlecht, jedoch in 1 N Natronlauge gut löslich ist. Die Strukturcharakterisierung erfolgte über1H-NMR und MALDI-MS (siehe 3.2)
p-(4,6-Diamino-s-triazin-2-yl)aminophenylarsonige Säure (MEL, Melarsen Oxid)
Melarsen (25) wird analog der Darstellung von PYR (16) (siehe 2.1.1) durch Schwefeldioxid und Kaliumiodid zum Arsonigsäuredichlorid (26) reduziert und in situ durch Natriumhydrogencarbonat zu p-(4,6-Diamino -s-triazin-2-yl) aminophenylarsoniger Säure (14) (MEL, Melarsen Oxid) hydrolysiert (Abb. 28: Darstellung) [124, 125].
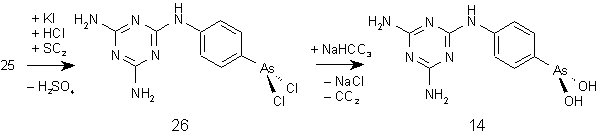
Abb. 28a: Darstellung von p-(4,6-Diamino-s-triazin-2-yl)aminophenylarsoniger Säure (Melarsen Oxid) (14).
Nach Umkristallisation mit Aktivkohle kristallisiert die Verbindung in Form langer, farbloser Nadeln. Die Elementaranalyse bestätigt ein Molekül Kristallwasser, das sich durch Erhitzen im Vakuum entfernen läßt [124].
Das 1H-NMR Spektrum von14 (Abb. 28 A) zeigt zwei Dupletts bei 7.46 und 7.86 ppm für die aromatischen Protonen, zwei Singuletts bei 6.34 und 6.60 ppm für die beiden Aminogruppen bzw. dem NH-Proton und ein Duplett bei 8.98 ppm für die OH-Protonen. Die weiteren Signale geringerer Intensität lassen auf geringe Anteile des zugehörigen oligomeren Anhydrids schließen, denn nach Zugabe von D2O verlieren nicht nur die NH- und OH-Signale wegen H/D-Austauschprozessen an Intensität, auch die Signale der aromatischen Protonen werden einheitlicher. (Abb. 28 B). Das läßt darauf schließen, daß durch Zugabe von Wasser (in Form von D2O) das Gleichgewicht (sieheAbb. 4) weiter auf die Seite der freien Arsonigen Säure verschoben wird.
Das FAB-Massenspektrum von 14 in Glycerin-Matrix (siehe Abb. 29) zeigt - analog zu PYR (16) - als Basispeak den zyklischen Arsonigsäurediester mit einem Molekül Glycerin (m/z 367). Mit geringeren Intensitäten finden sich der offenkettige Glycerindiester (m/z 459) und höhermolekulare kovalente Addukte aus zwei Molekülen Glycerin mit zwei (m/z 733) bzw. drei (m/z 1099) Molekülen Melarsen Oxid, sowie Glycerin-Cluster (m/z 825 und 917). Des weiteren ist das monomere Anhydrid (m/z 293) zu erkennen, jedoch nicht die freie Arsonige Säure (m/z 311).
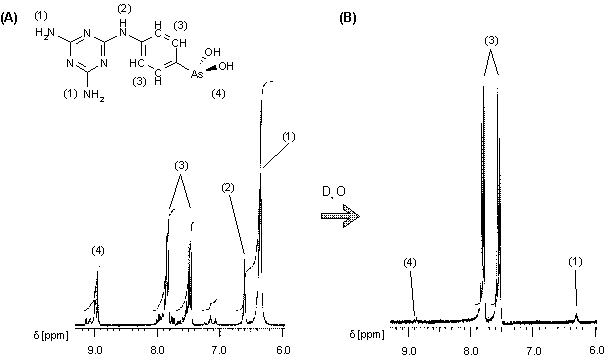
Abb. 28b:1H-NMR Spektrum von Melarsen Oxid (14) in d6-DMSO, intern mit TMS kalibriert. Dargestellt ist der aromatische Bereich (A) vor (B) nach Zugabe von D2O. Durch H/D-Austauschprozesse verlieren die NH-/OH-Signale an Intensität und die aromatischen Protonensignale werden einheitlicher.

Abb. 29: FAB-Massenspektrum von Melarsen Oxid (14) (Matrix: Glycerin / DMSO / AcOH 1:1:1).
Das MALDI-Massenspektrum von HPLC-gereinigtem MEL (14) (siehe Abb. 30) zeigt neben dem Anhydrid (m/z 293), der freien Säure (m/z 311), das dimere (m/z 585) und trimere (m/z 877) zyklische Anhydrid, HCCA-Mono- und Diester (m/z 482 bzw. 671), ähnlich den Glycerin-Addukten im FAB-MS (siehe Abb. 29). Der Peak bei m/z 477 kann dem dimeren Anhydrid, dem ein Triazinring fehlt (siehe vorgeschlagene Struktur in Abb. 31), zugeordnet werden. Die Spaltung der NH-Brücke könnte während der (sauren) Probenkristallisation auf dem MALDI-Target und / oder während der Desorption erfolgen.
Das ESI-Massenspektrum (siehe Abb. 31) einer Lösung von MEL (14) in MeOH /H2O 9:1 zeigt eindeutig, daß Melarsen Oxid überwiegend als Monomer in Form des Anhydrids (m/z 293) bzw. der freien Arsonige Säure (m/z 311) vorliegt. Die hier zusätzlich vorhanden Mono- und Dimethylester (m/z 325 und 339) resultieren aus dem verwendeten Lösungsmittelgemisch. Nur zu einem geringen Anteil liegt das zyklische dimere oder trimere Anhydrid (m/z 585 bzw. 877) vor. Auch hier scheint eine Spaltung der NH-Brücke in Melarsen Oxid unter sauren Bedingungen möglich, da der Peak m/z 477 dem dimeren Anhydrid, dem ein Triazinring fehlt, zugeordnet werden kann (siehe Abb. 31).
Die Gegenüberstellung der einzelnen MS-Methoden zeigt, daß die ESI-MS am besten die monomeren oder oligomeren Strukturen der Arsonigen Säuren wiedergibt. Sowohl FAB-MS, als auch MALDI-MS zeigen dagegen hauptsächlich Addukte mit der jeweilig verwendeten Matrix.
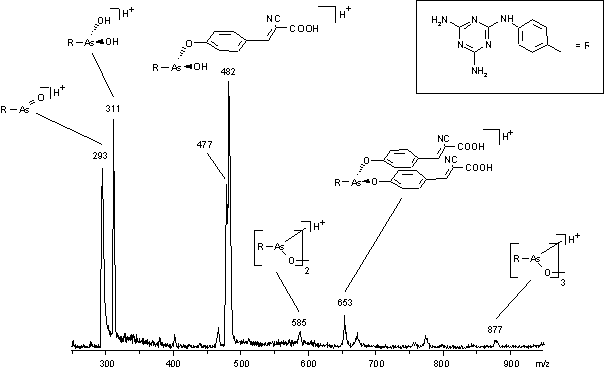
Abb. 30: MALDI-Massenspektrum von Melarsen Oxid (14) (Matrix: HCCA).

Abb. 31: ESI-Massenspektrum von Melarsen Oxid (14) in 2% AcOH / MeOH 9:1 (cmel = 100 mM).
2.1.3 p-Aminophenylarsonige Säure (A-PAA)
Die Synthese und Charakterisierung der p-Aminophenylarsonigen Säure (15) (A-PAA) (Abb. 18) ist in [119] beschrieben. Die Röntgenstruktur zeigt, daß A-PAA (15) in fester Form als Arsonige Säure vorliegt. Zusätzlich enthält die Struktur ein Molekül Kristallwasser, das die einzelnen A-PAA Moleküle über Wasserstoffbrücken stabilisiert [126]. Ähnliche kristallwasserhaltige Strukturen sind auch für die übrigen Derivate der Arsonigen Säure zu vermuten, sowie aus den Elementaranalysen zu schließen.
2.1.4 UV-spektroskopische Charakterisierung und Löslichkeitsverhalten der Arsonigsäure-Derivate
Die Löslichkeit von MEL, PYR und A-PAA in organischen Lösungsmitteln ist deutlich größer als die von PAA (sieheTab. 4). Jedoch sind Lösungen von MEL und A-PAA mit der in Tab. 4 angegebenen Konzentration nicht über längere Zeit stabil. Nach kurzer Zeit (MEL: max. 30 min, A-PAA: 1-2 h) fallen die Verbindungen in Form eines farblosen, amorphen Niederschlag aus. Vermutlich reagieren die freien Arsonigen Säuren in (fast) wasserfreier Umgebung zu den oligomeren Anhydriden, die bedeutend schlechter löslich sind und auch nicht durch Erwärmen wieder in Lösung gehen [119]. Dieser Prozeß wird im weiteren als 'altern' bezeichnet.
Tab. 4: Maximale Löslichkeit der Arsonigen Säuren in organischen Lösungsmitteln und wäßrigen Puffersystemen.
|
Lösungsmittel |
PAA [mol l-1] |
PYR [mol l-1] |
A-PAA [mol l-1] |
MEL [mol l-1] |
|
DMSO |
0.01 |
0.5 (a) |
0.5 |
0.35 |
|
MeOH |
n.b. |
> 0.5 |
> 0.5(b) 0.1 (c) |
> 0.35 (b)< 0.05 (c) |
|
MeOH/H2O 9:1 |
n.b. |
> 0.3 |
> 0.4 (b)0.4 (c) |
0.35 (b)0.1 (c) |
|
wäßrige Puffer (pH 2-8) |
n.b. |
n.b. |
n.b. |
pH 8: 0.2 10-3pH 2: 0.4 10-3 |
(Anmerkungen: (a) Lösung polymerisiert nach ca. vier Wochen; (b) Lösung frisch angesetzt; (c) gealterte Lösung, n.b. = nicht bestimmt)
Durch Zugeben von 10 % (v/v) Wasser zu frisch bereiteten Lösungen können diese stabilisiert werden, d.h. man stabilisiert das Gleichgewicht (vgl. Abb. 4) auf der Seite der Arsonigen Säure. So sind 0.1 M MEL- bzw. 0.4 M A-PAA-Lösungen in MeOH / H2O 9:1 mindestens 1 Woche bei 4 C lagerbar. PYR hingegen geht als zyklisches, oligomeres Anhydrid in Lösung (vgl.Abb. 25) und bleibt in dieser Form in Lösung mindestens vier Wochen stabil.
Der "Alterungsprozeß" einer A-PAA Lösung, die bereits eine Woche bei 4 C aufbewahrt wurde und aus der A-PAA z. T. wieder ausgefallen war, wurde mittels ESI-MS (siehe Abb. 32) untersucht. Das Spektrum unterscheidet sich deutlich zu PYR (Abb. 25) oder MEL (siehe Abb. 31). Als intensivstes Signal findet man das monomere Anhydrid (m/z 184), weniger intensiv sind die Signale für die freien Arsonige Säure (m/z 202) und das zyklische dimere Anhydrid (m/z 367). Höhermolekulare oligomere Anhydride sind nicht erkennbar, jedoch erkennt man mehrere Serien von Molekülionen mit einen Masseninkrement von m = 1831, also von molekularen Clustern, die jeweils eine monomere Anhydrideinheit mehr enthalten. Ein Startpunkt der Oligomerisierung könnte der Peak bei m/z 258 sein. Außerdem finden sich Masseninkremente von m = 141, die auf Methylester hinweisen. Vermutlich liegen keine zyklischen, sondern lineare Oligomere vor, wie auch schon durch EI-Massenspektren gezeigt wurde [119]. Das MALDI-Massenspektrum (Matrix: DHB oder ACH) zeigt für 150 m/z 750 im wesentlichen identische Signale (siehe 3.2.5).
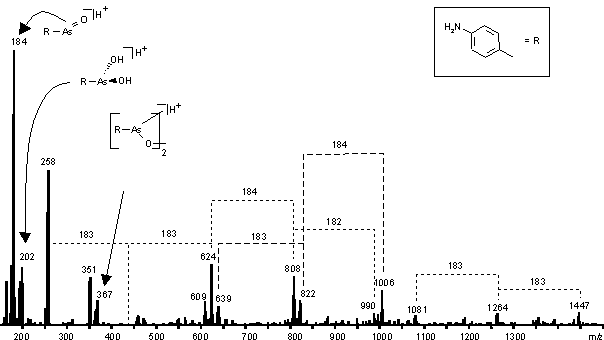
Abb. 32: ESI-MS von p-Aminophenylarsoniger Säure (A-PAA) in 2% AcOH / MeOH 9:1 (ca-paa = 60 mM). Die Lösung (300 mM in MeOH / H2O 9:1) wurde vor der Messung eine Woche bei 4 C aufbewahrt. A-PAA war z. T. "gealtert" und aus der Lösung ausgefallen.
Die UV-Spektren von MEL, PYR und A-PAA in MeOH / H2O 9:1 sind inAbb. 33 dargestellt. Dabei zeigt MEL die langwelligste Absorption und den größten Extinktionskoeffizienten (siehe Tabelle in Abb. 33). Das Absorptionsmaximum für PAA konnte nicht bestimmt werden, da die Eigenabsorption von DMSO das Absorptionsmaximum von PAA verdeckt und die Löslichkeit in Methanol zu gering war. Die exakte Bestimmung des molaren dekadischen Extinktionskoeffizienten ist wegen den unbekannten Gleichgewichten zwischen der freien Arsonigen Säure und den oligomeren Anhydriden (s.o.) erschwert. Gerade wegen der langwelligsten Absorption und dem hohen Extinktionskoeffizient eignet sich MEL - bei guter Löslichkeit - für UV-spektroskopische Untersuchungen der selektiven Proteinmodifizierung, denn Beeinflussungen durch Lösungsmittel und Proteineigenabsorption spielen bei 280 nm eine untergeordnete Rolle.
Des weiteren wurde das Absorptionsmaximum und der Extinktionskoeffizient von MEL in wäßrigen Puffersystemen bestimmt, die zur Proteinmodifikation Anwendung finden (siehe 3.3.1). Mit abnehmenden pH-Wert zeigt sich ein deutlich hypochromer Effekt (Abschwächung der Absorption), während das Absorptionsmaximum nahezu konstant bleibt (![]() max = 273-275). Ab pH=4 zeigt sich eine Schulter bei 247 nm, die vermutlich auf der Protonierung des Triazinrings beruht. Das Absorptionsmaximum wird in der Literatur [69] mit
max = 273-275). Ab pH=4 zeigt sich eine Schulter bei 247 nm, die vermutlich auf der Protonierung des Triazinrings beruht. Das Absorptionsmaximum wird in der Literatur [69] mit ![]() max = 272 nm, ein Wert für den Extinktionskoeffizienten dagegen nicht angegeben.
max = 272 nm, ein Wert für den Extinktionskoeffizienten dagegen nicht angegeben.
|
||||||||||||||||
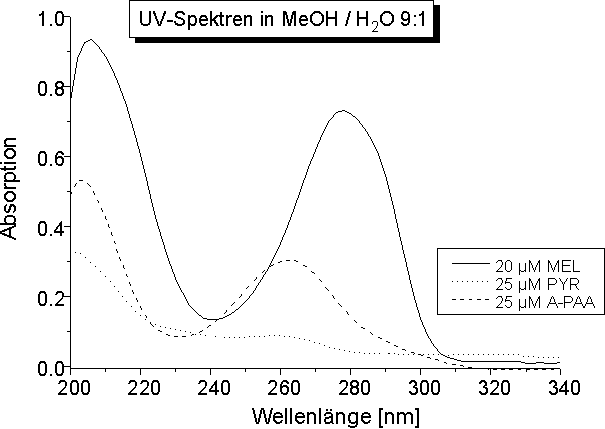
Abb. 33: UV-Spektren von 20 µM MEL, 25 µM PYR und 25 µM A-PAA im organischen Lösungsmittelsystem MeOH / H2O 9:1. Die Tabelle zeigt die dazugehörigen experimentell bestimmten Absorptionsmaxima und Extinktionskoeffizienten.
|
|
|
|
|
2.2 UV-spektroskopische Untersuchungen zum Reaktionsmechanismus der Modifizierung von Bis-Thiolen mit Melarsen Oxid |
|
|
1.5 Zielsetzung und Aufgabenstellungen der Arbeit |
Diese Seite wurde erstellt am 15. März 1998. Letzte Änderungen am 30. Juni 1998 durchPeter Happersberger.