|
|
|
|
|
2.7 Charakterisierung der in vitro Rückfaltungsreaktion von rhM-CSF |
|
|
2.6 Selektive chemische Modifizierung von Modellproteinen mit Melarsenoxid und massenspektrometrische Charakterisierung der Reaktionsprodukte
2.6.1 Umsetzung von Dicysteinylproteinen mit Melarsen Oxid
Als ein erstes Modellsystem wurde das Lungensurfactant Protein SP-C gewählt, da es wie das Peptid SP-C(1-14) (siehe 2.3.1) zwei vicinale Cysteine in reduzierter Form enthält. Reifes, humanes SP-C entsteht aus einem 21 kD Vorläuferprotein, besitzt 33-35 Aminosäuren und ist äußerst hydrophob [5].Abb. 65 zeigt die Aminosäuresequenz von synthetisch dargestelltem humanen SP-C [138].
Abb. 65: Aminosäuresequenz von synthetischem, humanen Lungensurfactant Protein SP-C; die vicinalen Cysteinreste sind fett gedruckt
Wesentliche Strukturmerkmale für die biologische Funktion sind (a) der sehr hydrophobe C-terminalen Sequenzbereich (Valincluster, 12-34) und (b) die vicinalen Cysteinreste, die im reifen Protein als posttranslationale Strukturmodifikation jeweils einen Palmitoylrest aufweisen [5]. Das synthetische SP-C besitzt keine Palmitoylierungen der Cysteine. SP-C ist aufgrund seiner hohen Hydrophobizität nur in Lösungen mit hohem organischen Lösungsmittelanteil löslich. Außerdem neigt SP-C in schwach saurem Medium zur Aggregation [135]. Von M. Kussmann durchgeführte Umsetzungen von rekombinanten SP-C mit PAA im molares Verhältnis SP-C / PAA 1:20 in Lösungen mit 80% Isopropanolanteil führten nicht zum Reaktionsprodukt SP-C-PAA [44].
Synthetisches SP-C wurde daher in situ auf den Nitrocellulose-PD-MS-Target mit MEL im molaren Verhältnis SP-C / MEL 1:20 umgesetzt. SP-C und MEL waren in IPA / 1% AcOH 4:1, pH 4 gelöst. In Abb. 66 A ist das PD-Massenspektrum von synthetischem SP-C gezeigt. Neben dem protonierten Molekülion bei m/z 3550 erkennt man sequenzspezifische Fragmentierungen der A-Ionenserie (siehe Fragmentierungsschema inAbb. 53), durch die die Aminosäuresequenz 8-22 des C-terminalen Bereichs abdeckt wird [135]. Das in Abb. 66 B dargestellte PD-Massenspektrum des mit MEL umgesetzten SP-Cs zeigt neben geringen Mengen an nicht modifizierten SP-C (m/z 3350) hauptsächlich MEL-modifiziertes SP-C (SP-C-MEL, m/z 3824). Die sequenzspezifischen Fragmentionen ![]() (n = 11-18) bestätigen die Modifizierung im Sequenzbereich 1-11. Das Fragment
(n = 11-18) bestätigen die Modifizierung im Sequenzbereich 1-11. Das Fragment ![]() zeigt, daß die Modifizierung innerhalb der ersten sechs Aminosäuren vorliegt und bestätigt damit die Selektivität der MEL-Modifizierungsreaktion für Biscysteinylfunktionen (vgl. Aminosäuresequenz in Abb. 65).
zeigt, daß die Modifizierung innerhalb der ersten sechs Aminosäuren vorliegt und bestätigt damit die Selektivität der MEL-Modifizierungsreaktion für Biscysteinylfunktionen (vgl. Aminosäuresequenz in Abb. 65).
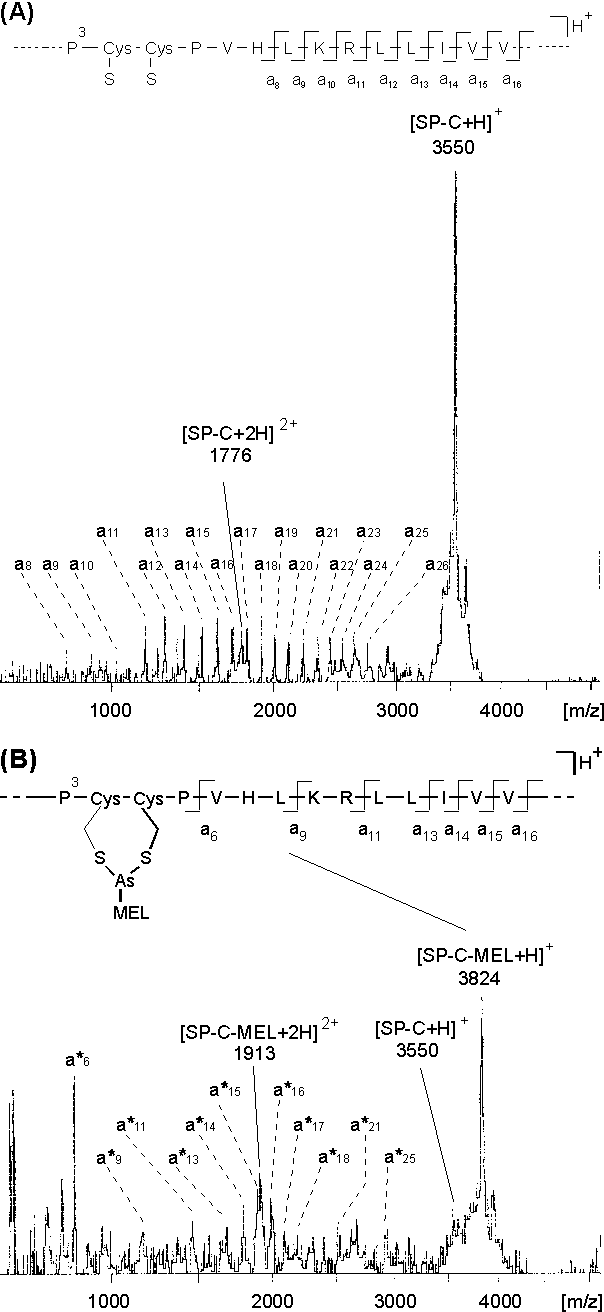
Abb. 66: (A) PD-Massenspektrum von synthetischen SP-C. Mit an sind sequenzspezifische Fragmentionen der A-Ionenserie gekennzeichnet. (B) PD-Massenspektrum von SP-C-MEL nach in situ-Modifizierung von SP-C mit MEL auf dem Nitrocellulose-PD-MS-Target. Die sequenzspezifischen Fragmentionen ![]() enthalten die MEL-Modifizierung.
enthalten die MEL-Modifizierung.
Um die in situ-Modifizierung zu bestätigen, wurde deshalb SP-C im Lösungsmittel IPA / 1% AcOH 4:1, pH 4 mit MEL im molaren Verhältnis SP-C / MEL 1:1 umgesetzt. Die Reaktion wurde UV-spektroskopisch verfolgt (siehe 2.2.2). Abb. 67 A zeigt den zeitlichen Verlauf der Absorptionsdifferenz OD299-267(t). Die Absorptionsdifferenz erreicht nach ca. 20 min ein Plateau. Damit verläuft die Reaktion von SP-C mit MEL ähnlich schnell wie die Umsetzung von Oxytocin mit MEL bei pH 4 (siehe Abb. 41). Die Reaktionslösung wurde ohne weitere Reinigungsschritte MALDI-massenspektrometrisch vermessen. Das in Abb. 67 B dargestellte MALDI-Massenspektrum zeigt das ein- und zweifach protonierte Molekülion von SP-C-MEL (m/z 3824 bzw. 1913), neben geringen Anteilen an nicht modifizierten SP-C (m/z 3550).

Abb. 67: Umsetzung von SP-C mit MEL in IPA / 1% AcOH 4:1, pH 4 im molaren Verhältnis 1:1. (A) Zeitlicher Verlauf der Absorptionsdifferenz ![]() OD299-267(t) für die Reaktion. (B) MALDI-Massenspektrum der Reaktionslösung nach 1h Reaktionsdauer. Das Massenspektrum wurde intern mit Insulin kalibriert (m/z 2868 für [I+2H]+ und 5735 für [I+H]+).
OD299-267(t) für die Reaktion. (B) MALDI-Massenspektrum der Reaktionslösung nach 1h Reaktionsdauer. Das Massenspektrum wurde intern mit Insulin kalibriert (m/z 2868 für [I+2H]+ und 5735 für [I+H]+).
2.6.2 Umsetzung von cystinylhaltigen Modellproteinen mit Melarsen Oxid und ihre Reinigung über HPLC
Die chemische Spezifität von Melarsen Oxid (MEL) für Biscysteinylfunktionen sollte nun an Modellproteinen untersucht werden, diemehrere Disulfidbrücken besitzen. Die Proteine Rinderpankreas-Trypsin-Inhibitor (bovine pancreatic trypsin inhibitor, BPTI; auch Aprotonin) und Insulin besitzen jeweils drei Disulfidbrücken, die vor der Reaktion mit MEL reduziert wurden.
BPTI inhibiert im Pankreas die Serinprotease Trypsin und verhindert dadurch den Selbstverdau des sekretorischen Organs. Das aus 58 Aminosäuren bestehende BPTI (MW = 6511.5 kD) besitzt Disulfidbrücken zwischen den Cysteinen Cys30-Cys51, Cys5-Cys55 und Cys14-Cys38. Die Aminosäuresequenz und Röntgenstruktur von BPTI (Auflösung 1.1 Å) zeigt Abb. 9 [139]. Der Rückfaltungsweg (folding pathway) wurde von Creighton et al. durch die Charakterisierung der Disulfidstrukturen der Faltungsintermediate sehr genau untersucht. Davon leitete er die Abfolge der Disulfidbrückenbildung während der oxidativen Rückfaltung ab [140, 32]. Untersuchungen von Weissman zeigen, daß dabei nur Intermediate mit nativen Disulfidbrücken eine wesentliche Rolle spielen [141]. Schematisch läßt sich der Faltungsweg von BPTI zusammenfassen:
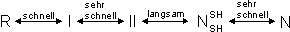
R repräsentiert das reduzierte und denaturierte BPTI, während N für das native Protein steht. I und II entsprechen Faltungsintermediaten mit einer bzw. zwei Disulfidbrücken. Das Faltungsintermediat ![]() enthält zwei der drei Disulfidbrücken des nativen Proteins (Cys30-Cys51, Cys5-Cys55). Vermutlich besitzt
enthält zwei der drei Disulfidbrücken des nativen Proteins (Cys30-Cys51, Cys5-Cys55). Vermutlich besitzt ![]() eine ähnliche Konformation wie natives BPTI (native-like), da es eine ähnlich hohe biologisch Aktivität wie BPTI besitzt. Der langsamste Schritt sowohl bei der oxidativen Rückfaltung, als auch bei der reduktive Entfaltung (reductive unfolding) ist die Gleichgewichtseinstellung zwischen II und
eine ähnliche Konformation wie natives BPTI (native-like), da es eine ähnlich hohe biologisch Aktivität wie BPTI besitzt. Der langsamste Schritt sowohl bei der oxidativen Rückfaltung, als auch bei der reduktive Entfaltung (reductive unfolding) ist die Gleichgewichtseinstellung zwischen II und ![]() . Damit ist das native Faltungsintermediat
. Damit ist das native Faltungsintermediat ![]() kinetisch äußerst stabil. Die Halbwertszeit für die reduktive Entfaltung (
kinetisch äußerst stabil. Die Halbwertszeit für die reduktive Entfaltung (![]() --> II) in Gegenwart von 1-50 mM DTT bei 25 C, pH 8.7 beträgt 20 h [33, 142, 27]. Die bevorzugte Spaltung der Disulfidbrücke Cys14-Cys30 erklärt sich aus der guten Oberflächenzugänglichkeit der Cysteine durch das Reduktionsmittel (siehe Abb. 68), während die restlichen Disulfidbrücken im Innern der Struktur verborgen liegen und somit kaum dem Reduktionsmittel zugänglich sind. In dieser Arbeit wurde sowohl teilweise wie auch vollständig reduziertes BPTI mit MEL umgesetzt.
--> II) in Gegenwart von 1-50 mM DTT bei 25 C, pH 8.7 beträgt 20 h [33, 142, 27]. Die bevorzugte Spaltung der Disulfidbrücke Cys14-Cys30 erklärt sich aus der guten Oberflächenzugänglichkeit der Cysteine durch das Reduktionsmittel (siehe Abb. 68), während die restlichen Disulfidbrücken im Innern der Struktur verborgen liegen und somit kaum dem Reduktionsmittel zugänglich sind. In dieser Arbeit wurde sowohl teilweise wie auch vollständig reduziertes BPTI mit MEL umgesetzt.
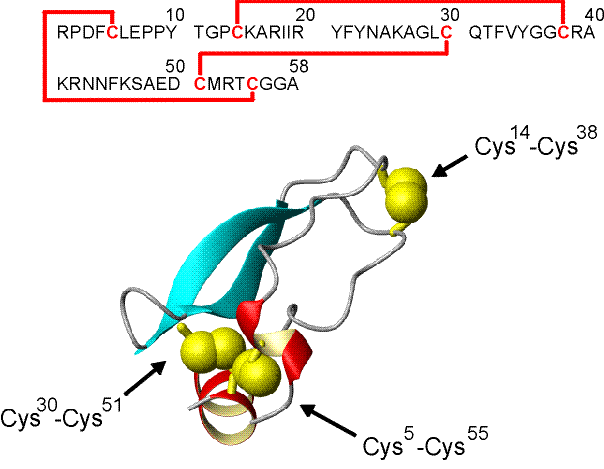
Abb. 68: Aminosäuresequenz und Röntgenstruktur von Bovinem Rinderpankreas-Trypsin-Inhibitor (BPTI). Die Disulfidbrücken Cys30-Cys51, Cys5-Cys55 und Cys14-Cys38, sowie der C- und N-Terminus sind gekennzeichnet.
Die partielle Reduktion von BPTI erfolgte im NH4HCO3-Puffer bei pH 8.2 mit TCEP im molaren Verhältnis BPTI / TCEP von 1:60 (siehe 3.4.1). Ein Aliquot der Lösung wurde mit IAA umgesetzt und MALDI-massenspektrometrisch charakterisiert. Das MALDI-Massenspektrum zeigte nahezu ausschließlich zweifach carboxamidomethyliertes BPTI. Nicht modifiziertes und sechsfach carboxamidomethyliertes BPTI lag nur zu geringen Anteilen vor. Das teilweise reduzierte BPTI wurde bei pH 8.2 (NH4HCO3-Puffer) mit MEL im molaren Verhältnis 1 zu 4 umgesetzt und die Reaktion UV-spektroskopisch verfolgt. Der zeitliche Verlauf der Absorptionsdifferenz ![]() OD299-267(t) (Abb. 69 A) zeigt, daß die Reaktion nach maximal 30 s beendet ist. Das MALDI-Massenspektrum (Abb. 69 B) der Reaktionslösung unmittelbar nach Zugabe von BPTI zur MEL-Lösung (innerhalb der 30 s) zeigt im wesentlichen die zweifach und einfach protonierten Molekülionen (m/z 3395 und 6789) des Reaktionsproduktes von einem Molekül BPTI und einem Molekül MEL (BPTI-MEL). Daneben finden sich Signale für BPTI (m/z 6512) und von dreifach MEL-modifizierten BPTI (7341, BPTI-3 MEL). Sowohl Signalintensitäten als auch die Art der Reaktionsprodukte stimmen sehr gut mit den erhaltenen Ergebnissen der IAA-Alkylierung überein. Die Reaktionslösungen der IAA-Alkylierung und MEL-Modifizierung wurden mittels Ultrafiltration (siehe 3.6.2) aufkonzentriert und, um die Hydrolyse (siehe 2.4.2) des BPTI-MEL-Addukts zu minimieren, auf pH 4 umgepuffert. Proteolytische Abbaureaktionen bei pH 4 mit V8-Protease ergaben für das zweifach carboxamidomethyliertes BPTI und für BPTI-MEL keine Peptide.
OD299-267(t) (Abb. 69 A) zeigt, daß die Reaktion nach maximal 30 s beendet ist. Das MALDI-Massenspektrum (Abb. 69 B) der Reaktionslösung unmittelbar nach Zugabe von BPTI zur MEL-Lösung (innerhalb der 30 s) zeigt im wesentlichen die zweifach und einfach protonierten Molekülionen (m/z 3395 und 6789) des Reaktionsproduktes von einem Molekül BPTI und einem Molekül MEL (BPTI-MEL). Daneben finden sich Signale für BPTI (m/z 6512) und von dreifach MEL-modifizierten BPTI (7341, BPTI-3 MEL). Sowohl Signalintensitäten als auch die Art der Reaktionsprodukte stimmen sehr gut mit den erhaltenen Ergebnissen der IAA-Alkylierung überein. Die Reaktionslösungen der IAA-Alkylierung und MEL-Modifizierung wurden mittels Ultrafiltration (siehe 3.6.2) aufkonzentriert und, um die Hydrolyse (siehe 2.4.2) des BPTI-MEL-Addukts zu minimieren, auf pH 4 umgepuffert. Proteolytische Abbaureaktionen bei pH 4 mit V8-Protease ergaben für das zweifach carboxamidomethyliertes BPTI und für BPTI-MEL keine Peptide. 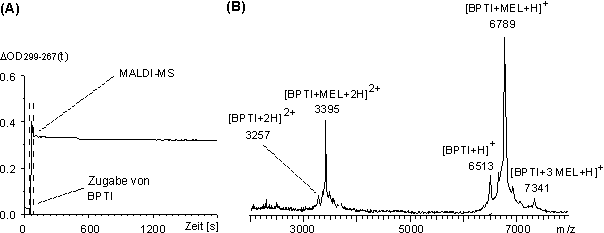
Abb. 69: Umsetzung von partiell reduzierten BPTI mit MEL im NH4HCO3-Puffer bei pH 8.2 im molaren Verhältnis BPTI / MEL 1:4. (A) Zeitlicher Verlauf der Absorptionsdifferenz OD299-267(t).(B) MALDI-Massenspektrum der Reaktionslösung nach ca. 30 s Reaktionszeit.
Die vollständige Reduktion von BPTI unter nicht denaturierenden Bedingungen gelingt innerhalb von Tagen bei Raumtemperatur [27]. Um die Reduktion zu beschleunigen wurden verschiedenen Reduktionsbedingungen ausgetestet (siehe 3.4.1). Der Nachweis der Reduktion erfolgte durch Umsetzung mit IAA und MALDI-MS-Analyse der Umsetzungsprodukte. Vollständig reduziertes BPTI - durch Reduktion unter denaturierenden Bedingungen - wurde im NaH2PO4-Puffer (pH 8.5 ) in Gegenwart von 4.5 M Harnstoff mit MEL im molaren Verhältnis BPTI / MEL 1:4 umgesetzt. Harnstoff wurde verwendet, um die Präzipitation von mehrfach MEL-modifizierten BPTI-Derivate zu minimieren. Die Reaktion wurde UV-spektroskopisch verfolgt; die Absorptionsdifferenz ![]() OD299-267(t) ist in Abb. 70 A dargestellt.
OD299-267(t) ist in Abb. 70 A dargestellt. ![]() OD299-267(t) erreicht innerhalb des Zeitfensters von ca. 30 s zum Öffnen des UV-Photometers einen maximalen Betrag. Das MALDI-Massenspektrum in Abb. 70 B der Reaktionslösung nach ca. 1200 s zeigt eine heterogene Produktverteilung von ca. 70% dreifach (m/z 7341), 30% zweifach (m/z 7065) und mit sehr geringer Intensität auch einfach MEL-modifiziertem BPTI (m/z 6789). Dagegen zeigte das MALDI-Massenspektrum der eingesetzten BPTI-Lösung nach Umsetzung mit Iodacetamid nahezu ausschließlich sechsfach carboxamidomethyliertes BPTI. Die heterogene Produktverteilung begründet sich vermutlich in der Schwerlöslichkeit mehrfach MEL-modifizierter BPTI-Derivate, da Umsetzungen von vollständig reduzierten BPTI mit MEL in Gegenwart geringerer Harnstoffkonzentrationen (0.7 M) zeigten, daß zwei- und dreifach MEL-modifiziertes BPTI präzipitieren (Abnahme der MALDI-MS-Signalintensitäten im Verlauf der Reaktion). Die Trennung von zweifach und dreifach MEL-modifizierten BPTI durch C18-RP-HPLC gelang nicht, wie die MALDI-MS-Analyse der HPLC-Fraktionen zeigte. HPLC-Trennungen auf weniger hydrophoben HPLC-Säulen wurde nicht durchgeführt.
OD299-267(t) erreicht innerhalb des Zeitfensters von ca. 30 s zum Öffnen des UV-Photometers einen maximalen Betrag. Das MALDI-Massenspektrum in Abb. 70 B der Reaktionslösung nach ca. 1200 s zeigt eine heterogene Produktverteilung von ca. 70% dreifach (m/z 7341), 30% zweifach (m/z 7065) und mit sehr geringer Intensität auch einfach MEL-modifiziertem BPTI (m/z 6789). Dagegen zeigte das MALDI-Massenspektrum der eingesetzten BPTI-Lösung nach Umsetzung mit Iodacetamid nahezu ausschließlich sechsfach carboxamidomethyliertes BPTI. Die heterogene Produktverteilung begründet sich vermutlich in der Schwerlöslichkeit mehrfach MEL-modifizierter BPTI-Derivate, da Umsetzungen von vollständig reduzierten BPTI mit MEL in Gegenwart geringerer Harnstoffkonzentrationen (0.7 M) zeigten, daß zwei- und dreifach MEL-modifiziertes BPTI präzipitieren (Abnahme der MALDI-MS-Signalintensitäten im Verlauf der Reaktion). Die Trennung von zweifach und dreifach MEL-modifizierten BPTI durch C18-RP-HPLC gelang nicht, wie die MALDI-MS-Analyse der HPLC-Fraktionen zeigte. HPLC-Trennungen auf weniger hydrophoben HPLC-Säulen wurde nicht durchgeführt.

Abb. 70: Umsetzung von vollständig reduziertem BPTI mit MEL im NaH2PO4-Puffer (4.5 M Harnstoff) bei pH 8.5 im molaren Verhältnis BPTI / MEL 1:4. (A) Zeitlicher Verlauf der Absorptionsdifferenz ![]() OD299-267(t). (B) MALDI-Massenspektrum der Reaktionslösung nach t 1200 s.
OD299-267(t). (B) MALDI-Massenspektrum der Reaktionslösung nach t 1200 s.
Als weiteres Modellsystem wurde Insulin gewählt, das ebenfalls drei Disulfidbrücken enthält, jedoch aus zwei unterschiedlichen Ketten zusammengesetzt ist. Das Hormon Insulin wird als Reaktion auf einen hohen Glucosespiegel vom Pankreas ins Blut sezerniert. Es regt dadurch Muskeln, Leber- und Fettgewebe dazu an, Glucose in Form von Glucogen, Protein und Fett zu speichern. Insulin besteht aus zwei Polypeptidketten mit insgesamt 51 Aminosäuren (MW = 5733.6 D). Kette A (21 Aminosäuren) und Kette B (30 Aminosäuren) sind über zwei intermolekulare (interkatenäre) Disulfidbrücken der Cysteine Cys7a-Cys7b und Cys20a-Cys19b kovalent verknüpft. Die A-Kette enthält ferner eineintramolekulare (intrakatenäre) Disulfidbrücke über die Cysteine Cys6a-Cys11a [1]. Die Aminosäuresequenz von humanem Insulin ist in Abb. 71 dargestellt.
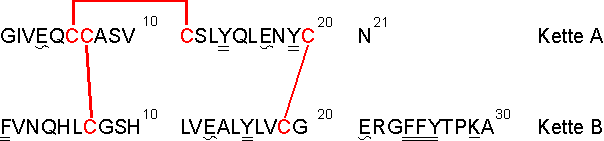
Abb. 71: Aminosäuresequenz von humanem Insulin. Die Disulfidbrücken Cys7a-Cys7b, Cys20a-Cys19b und Cys6a-Cys11a sind durch Striche gekennzeichnet. Cysteine sind fett gedruckt, einfach und zweifach unterstrichen sind tryptische und -chymotryptische, geschlängelt (~) unterstrichen sind V8-proteolytische Spaltstellen.
Nach vollständiger Reduktion erhält man zwei Polypeptidketten von denen die B-Kette zwei Cysteine (Cys7b und Cys19b), die durch 11 Aminosäuren voneinander getrennt sind enthält. Die Cysteine Cys6a und Cys7a der A-Kette liegen vicinal zueinander, während die Cysteine Cys11a und Cys20a durch drei bzw. sieben Aminosäuren vom nächsten Cystein getrennt sind. Insulin wurde als Modellprotein verwendet, da es eine Vielzahl an proteolytischen Spaltstellen (siehe Abb. 71) besitzt, so daß eine Identifizierung der genauen Position der MEL-Modifizierung möglich sein sollte. Außerdem interessierte, ob nach der MEL-Umsetzung MEL-verbrückte kovalente Homo- und/oder Heterodimere aus A- und/oder B-Kette gebildet werden (vgl. 2.4.1). Die vollständige Reduktion von Insulin erfolgte im NH4HCO3-Puffer bei pH 8.2 unter denaturierenden Bedingungen (3 M Harnstoff) mit TCEP im molaren Verhältnis Insulin / TCEP 1:60. Ein Aliquot der Reaktionslösung wurde mit Iodacetamid umgesetzt und ohne weitere Reinigungsschritte per MALDI-MS vermessen. Das MALDI-Massenspektrum inAbb. 72 A zeigt hauptsächlich die zweifach carboxamidomethylierte B-Kette (Ins-B-IAA2) bei m/z 3516 und die vierfach carboxamidomethylierte A-Kette (Ins-A-IAA4) bei m/z 2569, wodurch gezeigt wird, daß die Reduktion der Disulfidbrücken nahezu vollständig war. Geringe Signalintensitäten für die zweifach carboxamidomethylierte A-Kette (Ins-A-IAA2) bei m/z 2453 zeigen, daß möglicherweise die intramolekulare Disulfidbrücke zum geringen Teil nicht reduziert vorliegt. Die intramolekulare Disulfidbrücke Cys6a-Cys11a liegt im intakten Insulin verborgen und wird als letzte Disulfidbrücke reduziert [143]. Dagegen gelingt die Reduktion von Insulin in Lösung unter nicht denaturierenden Bedingungen nicht für Proteinkonzentrationen > 1 µg/µl, da die reduzierten Ketten präzipitieren. Auch bei der Modifizierung von reduziertem Insulin mit MEL im NH4HCO3-Puffer bei pH 8.2 fiel das Reaktionsprodukt selbst in Gegenwart von 6 M Harnstoff aus (vgl. nächster Abschnitt). Anzumerken ist noch, daß die Signale der A-Kette im Positivmodus aller MS-Methoden schlecht nachweisbar sind, da die A-Kette außer dem N-Terminus keine Protonierungsstellen besitzt. Im Negativmodus der MALDI-MS-Analyse zeigen A- und B-Kette etwa gleich Intensitäten.
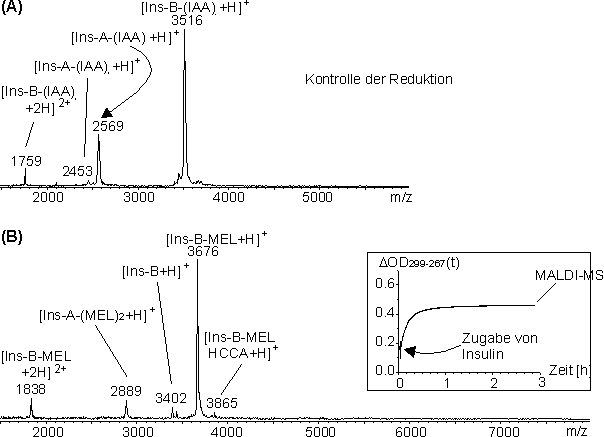
Abb. 72: MALDI-Massenspektren der Reaktionslösungen der Umsetzungen von reduziertem Insulin mit (A) Iodacetamid und (B) MEL im Glycin-Puffer bei pH 2.0 (5 M Harnstoff) nach t > 3 h. Als Insert ist der zeitlicher Verlauf der Absorptionsdifferenz ![]() OD299-267(t) dargestellt.
OD299-267(t) dargestellt.
Die Umsetzung von Insulin mit MEL erfolgte nach der Reduktion mit TCEP (s.o.) im Glycin-Puffer bei pH 2 in Gegenwart von 5 M Harnstoff. Das molare Verhältnis Insulin zu MEL betrug 12:40 (10-facher molarer Überschuß an MEL pro Biscysteinylfunktion). Die Reaktion wurde UV-spektroskopisch verfolgt. Der zeitliche Verlauf der berechneten Absorptionsdifferenz ![]() OD299-267(t) ist in Abb. 72 B (Insert) dargestellt. Die Reaktion verläuft ähnlich schnell wie die Umsetzung von Oxytocin mit MEL (siehe Abb. 41). Erst nach ca. 3 h erreicht
OD299-267(t) ist in Abb. 72 B (Insert) dargestellt. Die Reaktion verläuft ähnlich schnell wie die Umsetzung von Oxytocin mit MEL (siehe Abb. 41). Erst nach ca. 3 h erreicht ![]() OD299-267(t) einen maximalen Betrag. Das MALDI-Massenspektrum der nicht weiter gereinigten Reaktionslösung nach 3 h Reaktionszeit (Abb. 72 B) bestätigt die nahezu quantitative Modifizierung: Signale bei m/z 2889 entsprechen der zweifach MEL-modifizierten A-Kette (Ins-A-MEL2) und bei m/z 3676 der einfach MEL-modifizierten B-Kette (Ins-B-MEL). In geringen Anteilen liegt noch die unmodifizierte B-Kette (Ins-B) (m/z 3402) und das HCCA-Addukt von Ins-B-MEL (m/z 3865) vor (siehe 2.4.1). Es sind weder unspezifische Mehrfachmodifizierungen noch MEL-verbrückte Homo- oder Heterodimere der Polypeptidketten im hohen Massenbereich (m/z 4000 bis 8000) zu erkennen.
OD299-267(t) einen maximalen Betrag. Das MALDI-Massenspektrum der nicht weiter gereinigten Reaktionslösung nach 3 h Reaktionszeit (Abb. 72 B) bestätigt die nahezu quantitative Modifizierung: Signale bei m/z 2889 entsprechen der zweifach MEL-modifizierten A-Kette (Ins-A-MEL2) und bei m/z 3676 der einfach MEL-modifizierten B-Kette (Ins-B-MEL). In geringen Anteilen liegt noch die unmodifizierte B-Kette (Ins-B) (m/z 3402) und das HCCA-Addukt von Ins-B-MEL (m/z 3865) vor (siehe 2.4.1). Es sind weder unspezifische Mehrfachmodifizierungen noch MEL-verbrückte Homo- oder Heterodimere der Polypeptidketten im hohen Massenbereich (m/z 4000 bis 8000) zu erkennen.
Das Reaktionsgemisch wurde durch C18-RP-HPLC getrennt und die einzelnen Fraktionen MALDI-massenspektrometrisch untersucht. InAbb. 73 ist das HPLC-Chromatogramm (C) und die MALDI-Massenspektren von Fraktion 2 und 3 vor dem Lyophilisieren gezeigt.

Abb. 73: (A) und (B) MALDI-Massenspektren der HPLC-Fraktionen 2 (t = 18.6 min) und Fr. 3 (t = 22.4 min) des HPLC-Chromatogramms (C). Das MALDI-Massenspektrum der Fr. 4 (t = 23.2 min) ist identisch zu dem von Fr. 3. (C) HPLC-Chromatogramm der Reaktionslösung der Umsetzung von reduziertem Insulin mit MEL im Glycin-Puffer (5 M Harnstoff) bei pH 2 (siehe Abb. 72).
Das MALDI-Massenspektrum von Fr. 2 (Abb. 73 A) zeigt die einfach und zweifach protonierten Molekülionen von Ins-A-MEL2 (m/z 2889 und 1445), neben geringen Anteilen an Ins-A-MEL (m/z 2613) und dem HCCA-Addukt von Ins-A-MEL2 (m/z 3078). Die Signalintensitäten von einfach und zweifach modifizierter A-Kette stimmen sehr gut mit den Signalintensitäten der IAA-Alkylierung (Abb. 72 A) überein, so daß vermutlich die Disulfidbrücke Cys6a-Cys11a in Ins-A-MEL geschlossen vorliegt. Das MALDI-Massenspektrum von Fr. 3 (Abb. 73 B) zeigt nahezu ausschließlich Ins-B-MEL (3676). Die geringen Anteile an nicht modifizierter B-Kette begründen sich vermutlich durch die erneute säurekatalysierte Gleichgewichtseinstellung des Arsonigsäuredithioesters (vgl. 2.3.2). Bestätigt wird dies, durch die vollständige Abtrennung von nicht modifizierter B-Kette (Ins-B) aus der Reaktionslösung durch HPLC (Fr. 5 in Abb. 73 C). Die MALDI-Massenspektren von Fr, 3 und 4 sind identisch und entsprechen vermutlich den diastereomeren Reaktionsprodukten (siehe Abb. 8). Jedoch können die diastereomeren Reaktionsprodukte, sowie unterschiedliche Modifizierungsgrade der A-Kette durch C18-RP-HPLC nicht getrennt werden.
Zusammenfassend bestätigen die Umsetzungen der Modellproteine SP-C, BPTI und Insulin die chemisch Selektivität von MEL für Biscysteinylfunktionen im Protein. Es wurden keine unspezifischen Mehrfachmodifizierungen beobachtet. Probleme mit der Löslichkeit der Reaktionsprodukte bei pH 8 können durch Ausweichen in einen sauren pH-Bereich umgangen werden. Die Reaktionsprodukte zeigen im Sauren ähnliche gute Stabilitäten wie die des Vasopressin-MEL-Addukts.
2.6.3 Untersuchungen zum proteolytischen und chemischen Abbau von Vas-MEL und der MEL-modifizierten B-Kette des Insulins
Die proteolytische und chemische Spaltung der Polypeptidkette von chemisch modifizierten Proteinen mit bekannter Sequenz in definierte kleine Fragmente ist wesentliche Voraussetzung für die exakte Identifizierung der Modifizierungsposition in der Polypeptidkette. Der Spaltvorgang sollte vollständig und spezifisch sein, damit die Einzelsequenzen (bestimmt über das Molekulargewicht) der Peptidfragmente exakt innerhalb der gesamten Sequenz des Proteins zugeordnet werden können. Die Endoprotease Trypsin spaltet die C-terminalen Peptidbindungen der basischen Aminosäuren Lysin und Arginin. Die V8-Protease (Endopeptidase V8 aus Staphylococcus aureus), ebenfalls wie Trypsin eine Serinprotease, spaltet die C-terminalen Peptidbindungen der sauren Aminosäuren Glutaminsäure und Asparaginsäure. -Chymotrypsin besitzt eine deutlich geringere Spezifität als Trypsin und spaltet die C-terminalen Peptidbindungen v.a. der sperrigen, hydrophoben Aminosäuren Phenylalanin, Tyrosin und Tryptophan. Alle Proteasen besitzen einen optimalen pH-Bereich, in dem ihre Aktivität am höchsten ist. Für Trypsin und -Chymotrypsin liegt dieser bei ungefähr acht. V8-Protease zeigt eine vom verwendeten Puffer abhängige Substratspezifität. In 50 mM NH4OAc pH 4.0 oder 50 mM NH4HCO3 pH 7.8 spaltet V8 selektiv nach Glutaminsäure, während in 50 mM Natriumphosphat-Puffer pH 7.8 die Spaltung der Peptidbindung nach Glutaminsäure und Asparaginsäure erfolgt. Typische Reaktionszeiten für proteolytische Spaltungen sind 2-4 h [144].
Aufgrund der mäßigen Stabilität der Arsonigsäuredithioester im Basischen (siehe 2.4.2) ist die proteolytische Spaltung der mit MEL modifizierte Peptide / Proteine nicht unter Standardbedingungen durchführbar. Alternativen wären zum einen, die eingesetzte Enzymmenge zu erhöhen (von Enzym / Substrat (E/S) 1:50 auf 1:5), um somit die proteolytische Spaltung zu beschleunigen. Zum anderen stehen Proteasen mit optimalen pH-Bereichen im Sauren (V8, pH 4; Pepsin pH 2) zur Verfügung. Jedoch zeigt Pepsin eine nur sehr geringe Substratspezifität.
Erste proteolytische Abbaureaktionen wurde an HPLC-gereinigtem Vas-MEL (siehe 2.3.2) durchgeführt. Vasopressin besitzt eine tryptische (Arg8) und zwei -chymotryptische Spaltstellen (Tyr2 und Phe3, siehe Aminosäuresequenzen inAbb. 43). Lyophilisierte Aliquote von Vas-MEL wurden im NaH2PO4-Puffer (50 mM, pH 8.5) gelöst und mit Trypsin bzw. -Chymotrypsin im Verhältnis E/S 1:5 und bei 25 C inkubiert. Zusätzlich wurde Vasopressin (nicht reduziert) mit - Chymotrypsin unter den gleichen Bedingungen versetzt. Der zeitliche Verlauf der Proteolyse wurde MALDI-massenspektrometrisch verfolgt, um die Abbaureaktion mit der Hydrolysegeschwindigkeit zu vergleichen. Das MALDI-Massenspektrum des Trypsinabbaus nach 30 min (Abb. 74) zeigt Signale für die tryptischen Fragmente [Aminosäure 1-8] mit (m/z 1305) und ohne MEL-Modifizierung (m/z 1029). Die tryptische Spaltung der Polypeptidkette erfolgt selektiv und quantitativ. Die zyklische Struktur des Arsonigsäuredithioesters bleibt erhalten. Der relativ hohe Anteil an nicht modifizierten Vasopressin dürfte auf die partielle Hydrolyse des Vas-MEL-Addukts zurückzuführen sein.
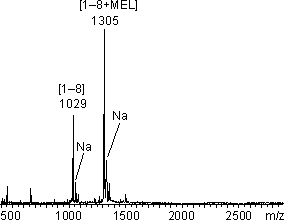
Abb. 74: MALDI-Massenspektrum des proteolytischen Abbaus von Vas-MEL mit Trypsin (E/S 1:5), 50 mM NaH2PO4 pH 8.5 nach 30 min.
Der -chymotryptische Abbau von Vasopressin zeigt auch nach 2 h Reaktionszeit keinerlei Spaltprodukte. Dagegen zeigt das MALDI-MS des -chymotryptischen Abbaus von Vas-MEL bereits nach 30 min (Abb. 75) eine Vielzahl an Signalen, die sich folgendermaßen unterteilen lassen: Gruppe (a) enthält die nicht proteolytisch gespaltenen Peptide Vas-MEL (m/z 1361) bzw. sein HCCA-Addukt (m/z 1550) und Vasopressin, das aus der Hydrolyse von Vas-MEL entstanden ist. In der Gruppe (b) sind die erwarteten -chymotryptischen Fragmente zusammengefaßt, die noch die MEL-Modifizierung enthalten So entspricht das Signal bei m/z 1379 der an der Aminosäure Tyr2 oder Phe3 einfach gespaltenen Polypeptidkette von Vas-MEL. Das deutlich weniger intensive Signal bei m/z 1232 kann dem Fragment mit der aus der Polypeptidkette abgespalteten Aminosäure Phe3 zugeordnet werden. Beide Fragmente entsprechen damit offenkettigen Arsonigsäuredithioestern. Zur Gruppe (c) zählen die -chymotryptischen Fragmente [3-9] (m/z 821) und [4-9] (m/z 674), die keine MEL-Modifizierung enthalten. Die Entstehung dieser Fragmente kann zum einen auf die -chymotryptischen Spaltung des nicht modifizierten und reduziert vorliegenden Vasopressins zurückgeführt werden, das durch Hydrolyse von Vas-MEL nachgebildet wird. Zum anderen ist die Hydrolyse der offenkettigen Arsonigsäuredithioester zur Arsonigen Säure und den entsprechenden Thiolen denkbar. In beiden Fällen enthalten die -chymotryptischen Fragmente [1-2], [1-3], [3-9] und [4-9] jeweils ein Cystein, das vermutlich reduziert vorliegt, denn die der Gruppe (d) zugeordneten Signale entsprechen offenkettigen Arsonigsäuredithioester aus MEL und den einzelnen -chymotryptischen Fragmenten. Die MEL-verbrückten Peptide entsprechen nicht der ursprünglichen Aminosäuresequenz des Edukts Vas-MEL und könnten durch Ligandenaustausch oder Umesterungsreaktionen analog der Spaltung der As-S Bindung von Vas-MEL durch DTT (siehe 2.4.3) entstanden sein. In Abb. 75 sind die gefunden Signale bei m/z 844, 1138, 1621 und 1770 den entsprechenden Strukturen zugeordnet. Dabei entspricht die Schreibweise, z.B. von MEL(CY)2 dem offenkettigen Arsonigsäuredithioester aus MEL und dem cysteinhaltigen Peptidfragment [1-2]. Eine Ausnahme bildet das Signal bei m/z 967, das dem offenkettigen Arsonigsäuremonothioester aus MEL und [4-9] zugeordnet werden kann. Die Abspaltung der OH Gruppe (m/z 950) könnte darauf hinweisen, daß eine weitere nukleophile Gruppe des Partialpeptids [4-9] an das Arsenatom koordiniert, z.B. das N-Atom des Prolins oder Arginins. Der -chymotryptische Abbau von Vas-MEL ist nach 2h beendet.
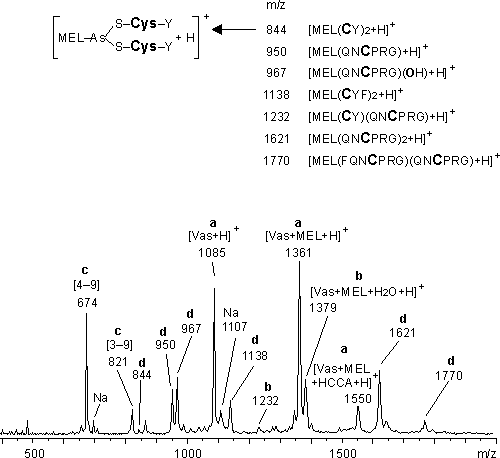
Abb. 75: MALDI-Massenspektrum des proteolytischen Abbaues von Vas-MEL mit -Chymotrypsin (E/S 1:5), 50 mM NaH2PO4 pH 8.5 nach 30 min. Die Zuordnung der Signale zu einzelnen Gruppen (a-d) erfolgt im Text. Die ergänzende Zuordnung der Signale ist in tabellarischer Form dargestellt. Fett und größer gedruckt sind die Cysteine der Partialpeptidketten, über deren S-Atom die Bindung zum Arsen erfolgt. Die gebildeten Produkte entsprechen offenkettigen Arsonigsäuredithioester.
Die über HPLC gereinigte MEL-modifizierte B-Kette des Insulins (Ins-B-MEL) (siehe 2.6.2) wurde ebenfalls proteolytisch abgebaut. Die Polypeptidkette besitzt zwei tryptische (Arg 22, Lys29), vier -chymotryptische (Tyr16, Phe24, Phe25, Tyr26) und zwei V8-proteolytische Spaltstellen (siehe Sequenz in Abb. 71). Aliquote an lyophilisiertem Ins-B-MEL wurden im NaH2PO4-Puffer (50 mM, pH 8.5) und mit Trypsin, -Chymotrypsin und V8-Protease (E/S 1:5) bei 25 C abgebaut. Gleiche Bedingungen wurden für den V8-Proteaseabbau bei pH 4 gewählt. Der Zeitverlauf der Proteolysen wurde MALDI-massenspektrometrisch verfolgt. Abb. 76 A zeigt das MALDI-Massenspektrum des Trypsinabbaus nach 25 min. Man erkennt Signale bei m/z 2763 für das MEL-modifizierte Partialpeptid [1-22+MEL] bzw. bei m/z 2487 für das Hydrolyseprodukt [1-22] und bei m/z 860 für [23-29]. Die -chymotryptische Aktivität des Trypsins führt zum Auftreten des Partialpeptid [1-16] bei m/z 1830. Der -Chymotrypsinabbau (MALDI-Massenspektrum, nach 25 min in Abb. 76 B) zeigt neben dem nicht abgebauten Ins-B-MEL (m/z 3676) und nicht modifizierter B-Kette (m/z 3402) die Peptide [17-24] (m/z 880), [17-25] (m/z 1027) und [1-16] (m/z1830), die jeweils ein Cystein enthalten,aber nicht mit MEL modifiziert sind. Das Signal bei m/z 3130 kann dem MEL-modifizierten Peptid [2-26+MEL] zugeordnet werden. Aufgrund der Massenauflösung des MALDI-MS kann zwischen dem Na-Addukt (m = 22) und der einfach hydrolysierten (-chymotryptisch gespaltenen), MEL-modifizierten B-Kette (m = 18) nicht unterschieden werden.
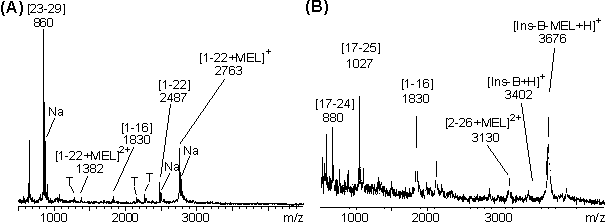
Abb. 76: MALDI-Massenspektren des proteolytischen Abbaues nach 25 min von Ins-B-MEL mit (A) Trypsin, (B) mit -Chymotrypsin Die proteolytischen Abbaureaktionen erfolgten im Verhältnis E/S 1:5 in 1 M Harnstoff, 50 mM NaH2PO4 pH 8.5. Mit 'Na' sind die Na-Addukte, mit 'T' Autoproteolyseprodukte gekennzeichnet. Die MALDI-Massenspektren (Abb. 77 A und B) des V8-Abbaus nach 25 min bei pH 8 und pH 4 zeigen die nicht MEL-modifizierten Peptide [1-13] (m/z 1483), [14-21] (m/z 867) und [22-30] (m/z 1087); damit wird die Sequenz der B-Kette vollständig abgedeckt. Im Gegensatz zu pH 8 ist der V8-Abbau bei pH 4 (auch nach 2 h) nicht vollständig (Abb. 77 B). Neben der nicht abgebauten B-Kette (Ins-B und Ins-B-MEL) lassen sich die Signale bei m/z 1935 und 2606 den Peptiden [14-30] und [1-21+MEL] zuordnen.
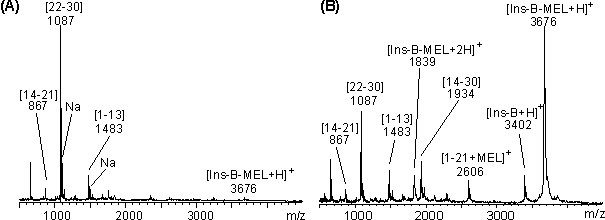
Abb. 77: MALDI-Massenspektren des proteolytischen Abbaus nach 25 min von Ins-B-MEL mit V8-Protease bei (A) pH 8 und(B) bei pH 4. Die proteolytischen Abbaureaktionen erfolgten im Verhältnis E/S 1:5 in 1 M Harnstoff, 50 mM NaH2PO4 pH 8.5. Mit 'Na' sind die Na-Addukte gekennzeichnet (wegen der Übersichtlichkeit nicht in (B)).
Die Tatsache, daß MEL-modifizierte proteolytische Fragmente nur dann beobachtet werden, wenn das Partialpeptid zwei Cysteine enthält, läßt vermuten, daß die nach der proteolytischen Spaltung vorliegenden offenkettigen Arsonigsäuredithioester aufgrund ihrer geringeren Stabilität (vgl. 1.2.3) sofort in die nicht modifizierten Partialpeptide hydrolysieren. Denkbar wäre auch, daß durch die MEL-Modifizierung die Polypeptidkette proteolyseresistent wird bzw. eine proteolytische Spaltung nur "links und rechts" der modifizierten Cysteine erfolgen kann. Die beobachteten proteolytischen Fragmente würden dann aus dem Abbau der nicht modifizierten B-Kette resultieren, die aufgrund der Hydrolyse von Ins-MEL ständig nachgebildet wird. Aus den vorhandenen Daten geht nicht hervor, welcher der beiden Mechanismen tatsächlich vorliegt. In Tab. 7 sind die gefundenen proteolytischen Fragmente für die einzelnen Proteasen zusammengefaßt. Die Ergebnisse stehen im Einklang mit Untersuchungen von C. Borchers an reduziertem Insulin [145].
Tab. 7: Massenspektrometrische Analyse und Zuordnung der proteolytischen Partialpeptide der MEL-modifizierten B-Kette des Insulins.
|
Protease / pH-Wert |
Peptid |
m/zgefunden |
m/z berechnet |
|
Trypsin / pH 8.5 |
[1-22] |
2486 |
2487 |
|
[1-22+MEL] |
2762 |
2763 |
|
|
[23-29] |
860 |
860 |
|
|
-Chymotrypsin /pH 8.5 |
[1-16] |
1833 |
1830 |
|
[17-24] |
881 |
880 |
|
|
[17-25] |
1029 |
1028 |
|
|
[2-26+MEL] |
3131 |
3130 |
|
|
V8-Protease / pH 8.5 |
[1-13] |
1483 |
1483 |
|
[14-21] |
868 |
867 |
|
|
[22-30] |
1087 |
1087 |
|
|
zusätzlich bei pH 4.0 |
[1-21+MEL] |
1934 |
1935 |
|
[14-30] |
2606 |
2606 |
Analoge Untersuchungen wurden für die MEL-modifizierte A-Kette durchgeführt. Wegen der geringeren Löslichkeit der MEL-modifizierten A-Kette in Vergleich zu Ins-B-MEL und wegen schlechteren Desorptionseigenschaften erhält man in den MALDI-Massenspektren nur wenig intensive Signale, die die Zuordnung der proteolytischen Fragmente erschweren.
Eine weitere Methode zur Zerlegung einer Polypeptidkette in Partialpeptide ist die Bromcyan-Spaltung. BrCN spaltet in saurer Lösung (pH 2) spezifisch und quantitativ die C-terminale Peptidbindung von Methioninresten unter Bildung eines Gemisches aus Peptidyl-Homoserin und -Homoserinlacton [146]. Um die Stabilität der MEL-Modifizierung unter BrCN-Abbaubedingungen zu untersuchen, wurde HPLC-gereinigtes, lyophilisiertes Vas-MEL (s.o.) in 0.1% TFA / 0.1% TFA in ACN 6:4 (pH 2) gelöst und mit BrCN im molaren Verhältnis von 1:1000 versetzt und bei 25 C inkubiert. Die MEL-Modifizierung ist in diesem Lösungsmittel (ohne BrCN) bei -20 C mindestens vier Wochen stabil. Das MALDI-Massenspektrum (nicht dargestellt) zeigt nach 30 min Reaktionszeit zu 75% und nach 1 h ausschließlich nicht modifiziertes Vasopressin. Trivalent organylsubstituierte Arsine addieren BrCN, dabei wird das As(III) zu As(V) oxidiert [147]. Im Fall des Vas-MEL führt die Reaktion vermutlich zur Hydrolyse in die freie Arsonsäure (Melarsen) und zum reduziert vorliegenden Vasopressin.
|
|
|
|
|
2.7 Charakterisierung der in vitro Rückfaltungsreaktion von rhM-CSF |
|
|
Diese Seite wurde erstellt am 15. März 1998. Letzte Änderungen am 30. Juni 1998 durchPeter Happersberger.