|
|
|
|
|
2.6 Selektive chemische Modifizierung von Modellproteinen mit Melarsenoxid und massenspektrometrische Charakterisierung der Reaktionsprodukte |
|
|
2.4 Untersuchungen zur Stabilität der Reaktionsprodukte von mit Melarsen Oxid modifizierten Modellpeptiden |
2.5 Anwendung der Arsonigen Säuren als Schutzgruppe für Biscysteinyl-Gruppierungen in Peptiden gegen Alkylierungsmittel
Es sollte nun die Stabilität der Arsonigsäuredithioester nicht an isolierten (HPLC-gereinigten) Proben, sondern in der Reaktionslösung untersucht werden. UV-spektroskopische Untersuchungen der in situ-Reduktion von Oxytocin (OT) in Gegenwart von MEL (siehe Abb. 45) zeigten eine leichte Abnahme der Absorptionsdifferenz ![]() OD299-267 nach ca. 1 h Reaktionszeit. Da die Reaktion in 50 mM NH4HCO3 pH 8.2 durchgeführt wurde liegt der Rückschluß nahe, daß analog zu den Untersuchungen an Vas-MEL, die Abnahme der Absorptionsdifferenz auf eine geringere Stabilität des OT-MEL-Addukts in diesem Puffer zurückzuführen ist (siehe 2.4.2). Insbesondere interessierte die Frage, ob die während der Modifizierungsreaktion ausgebildeten Arsonigsäuredithioester stabile Ringstrukturen ausbilden oder, ob (reversible) Ringöffnungs- und Ringschlußmechanismen vorliegen. Das reversible Gleichgewicht wäre UV-spektroskopisch nicht von stabilen Ringstrukturen zu unterscheiden. Die Ausbildung stabiler As-S-Bindungen ist besonders wichtig für die Charakterisierung von Faltungsintermediaten, da die Zuordnung der möglichen Disulfidbrücken in Proteinen über die jeweilig modifizierten Cysteinpaare erfolgen soll. Zur Untersuchung dieser Fragestellung sind cysteinselektive Modifizierungsreagenzien, wie Iodacetamid (IAA) oder 4-Vinylpyridin (VP) für den schwach alkalischen bis neutralen pH-Bereich geeignet. Im Sauren können 2-[(2-Pyridylmethyl)-sulfinyl]-1R-benzimidazole, die gemischte Disulfidaddukte bilden [44] oder Arsonigsäure-Derivate verwendet werden. Die Modifizierungsreagenzien würden im Fall des (reversiblen) Bindungsbruchs der As-S-Bindung mit der Thiolkomponente reagieren. Ein Rückbildung des zyklischen Arsonigsäuredithioesters wäre nicht möglich, so daß der offenkettige Monothioester vollständig hydrolysiert (Abb. 1). Voraussetzung ist, daß die Modifizierung der Thiolgruppe schneller erfolgt als die Rückbildung des zyklischen Arsonigsäuredithioesters.
OD299-267 nach ca. 1 h Reaktionszeit. Da die Reaktion in 50 mM NH4HCO3 pH 8.2 durchgeführt wurde liegt der Rückschluß nahe, daß analog zu den Untersuchungen an Vas-MEL, die Abnahme der Absorptionsdifferenz auf eine geringere Stabilität des OT-MEL-Addukts in diesem Puffer zurückzuführen ist (siehe 2.4.2). Insbesondere interessierte die Frage, ob die während der Modifizierungsreaktion ausgebildeten Arsonigsäuredithioester stabile Ringstrukturen ausbilden oder, ob (reversible) Ringöffnungs- und Ringschlußmechanismen vorliegen. Das reversible Gleichgewicht wäre UV-spektroskopisch nicht von stabilen Ringstrukturen zu unterscheiden. Die Ausbildung stabiler As-S-Bindungen ist besonders wichtig für die Charakterisierung von Faltungsintermediaten, da die Zuordnung der möglichen Disulfidbrücken in Proteinen über die jeweilig modifizierten Cysteinpaare erfolgen soll. Zur Untersuchung dieser Fragestellung sind cysteinselektive Modifizierungsreagenzien, wie Iodacetamid (IAA) oder 4-Vinylpyridin (VP) für den schwach alkalischen bis neutralen pH-Bereich geeignet. Im Sauren können 2-[(2-Pyridylmethyl)-sulfinyl]-1R-benzimidazole, die gemischte Disulfidaddukte bilden [44] oder Arsonigsäure-Derivate verwendet werden. Die Modifizierungsreagenzien würden im Fall des (reversiblen) Bindungsbruchs der As-S-Bindung mit der Thiolkomponente reagieren. Ein Rückbildung des zyklischen Arsonigsäuredithioesters wäre nicht möglich, so daß der offenkettige Monothioester vollständig hydrolysiert (Abb. 1). Voraussetzung ist, daß die Modifizierung der Thiolgruppe schneller erfolgt als die Rückbildung des zyklischen Arsonigsäuredithioesters.
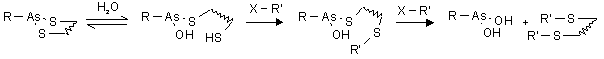
Abb. 60: Schematische Darstellung zum Nachweis eines möglichen Hydrolysegleichgewichts von Arsonigsäuredithioester durch cysteinselektive Modifizierungsreagenzien (R'-X).
Folgende Experimente wurden durchgeführt: MEL wurde im (I) NH4HCO3-Puffer bei pH 8.0 und (II) in NaH2PO4-Puffer bei pH 8.5 mit TCEP-reduziertem Oxytocin im molaren Verhältnis 4:3 umgesetzt. Zu diesen Reaktionslösungen und zur Kontrolle (gleiche Konzentrationen an reduziertem OT und identische Puffer ohne MEL) wurden dann in bestimmten Zeitabständen 4-Vinylpyridin zugeben, so daß molare Verhältnisse MEL / OT / VP 4:3:20 und 4:3:120 resultierten (siehe 3.4.3).
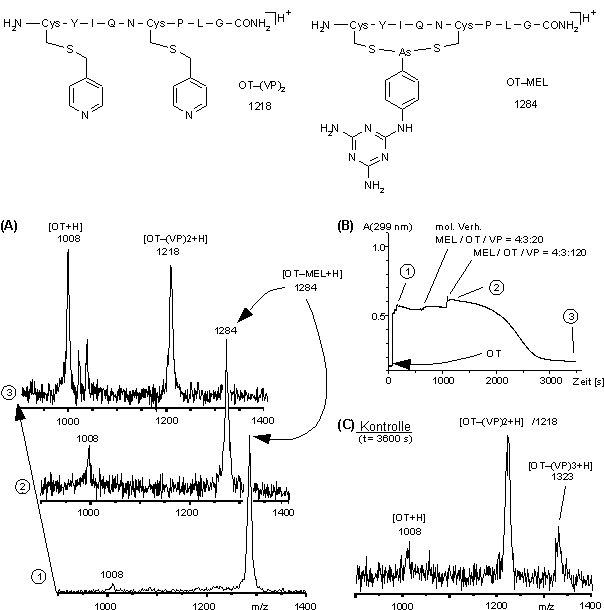
Abb. 61: Stabilität des OT-MEL-Addukts im NH4HCO3-Puffer bei pH 8.0 in Gegenwart des Alkylierungsmittels 4-Vinylpyridin im molaren Verhältnis OT / MEL / VP 4:3:20 und 4:3:120. (A) MALDI-Massenspektren vor und nach Zugabe von VP. Die Ziffern (1), (2) und (3) entsprechen den Zeitpunkten in (B). (B) Zeitlicher Verlauf der Absorption bei 299 nm. (C) MALDI-Massenspektrum der Kontrolle (OT und VP, ohne MEL) zum Zeitpunkt (3).
Aliquote der Reaktionslösungen MALDI-massenspektrometrisch vermessen. Die Reaktion wurde UV-spektroskopisch verfolgt. Aufgrund der Eigenabsorption von VP im Bereich von 220-280 nm ist die Berechnung der Absorptionsdifferenz OD299-267 (siehe 2.2.2) nicht möglich. Jedoch gibt die zeitliche Änderung der Absorption A299(t) bei 299 nm recht gut den zeitlichen Reaktionsverlauf (OD299-267(t)) wieder (siehe Abb. 40). InAbb. 61 sind die Ergebnisse für den NH4HCO3-Puffer zusammengefaßt. Abb. 61 B zeigt A299(t). Das MALDI-Massenspektrum (1) in Abb. 61 A zeigt neben geringen Mengen an OT (m/z 1008), die nahezu quantitative Umsetzung zum OT-MEL-Addukt (m/z 1284). Das MALDI-Massenspektrum (2) (Abb. 61 A) zeigt kurz nach VP-Zugabe (molares Verhältnis Cystein / VP 1:20) noch keine Änderung zum Spektrum (1). Jedoch zeigt die abnehmende Absorption A299(t) nach VP-Zugabe die Rückbildung von freiem MEL. Das MALDI-Massenspektrum (3) (Abb. 61 A) zeigt Signale für nicht modifiziertes (disulfidverbrücktes) OT (m/z 1008) und zweifach S-pyridylethyliertes OT (m/z 1218), aber nicht für das OT-MEL-Addukt. Die vollständige Hydrolyse von OT-MEL in Gegenwart von VP bestätigt die geringe Stabilität der Arsonigsäuredithioester im NH4HCO3-Puffer (pH 8.0). Das MALDI-Massenspektrum (Abb. 61 C) der Kontrollreaktion zum Zeitpunkt (3) (Abb. 61 A) zeigt zweifach (1218) und dreifach (1323) S-pyridylethyliertes OT. Neben den beiden Cysteinen wurde vermutlich der N-Terminus alkyliert.
In Abb. 62 sind die Ergebnisse für den NaH2PO4-Puffer zusammengefaßt. Auch nach Zugabe von VP im molaren Verhältnis Cystein / VP 1:20 erfolgt keine Abnahme von A299(t) (Abb. 62 A). Die MALDI-Massenspektren (Abb. 62 B) vor (1) und nach (2) Zugabe von VP sind praktisch identisch und zeigen nur den protonierten Oxytocin- melarsonigsäuredithioester (m/z 1284) bzw. das Na-Addukt (m/z 1306) davon. Das MALDI-Massenspektrum (nicht dargestellt) der Kontrolle zum Zeitpunkt (2) zeigte einfach und zweifach S-pyridylethyliertes Oxytocin im Verhältnis von ca. 1:2. Die Arsonigsäuredithioester sind also in nicht-nukleophilen Puffersystemen (siehe 2.4.2) in Gegenwart von cysteinselektiven Modifizierungsreagenzien deutlich stabiler, als in nukleophilen Puffersystemen.
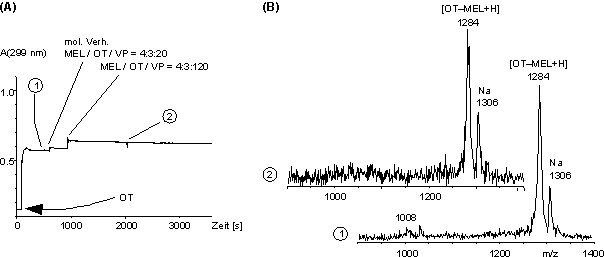
Abb. 62: Stabilität des OT-MEL-Addukts im NaH2PO4 bei pH 8.5 in Gegenwart des Alkylierungsmittels 4-Vinylpyridin im molaren Verhältnis OT / MEL / VP 4:3:20 und 4:3:120. (A) Zeitlicher Verlauf der Absorption bei 299 nm. (B) MALDI-Massenspektren vor und nach Zugabe von VP. Die Ziffern (1) und (2) entsprechen den Zeitpunkten in (A).
Somit sollte es möglich sein MEL als Schutzgruppe für Cysteine gegen andere cysteinselektive Modifizierungsreagenzien einzusetzen. Die wesentliche Eigenschaft einer Schutzgruppe ist ihr reversibles Abspalten, so daß die geschützte Funktion in ihrer ursprünglichen (chemischen) Form vorliegt. Geeignete Entschützungsreagenzien wären im Fall der Arsonigen Säuren Dithiole, z.B. 1,4-Dithiothreitol (DDT).
Im Folgenden sollte die Eignung von MEL als Schutzgruppe zur anschließenden Alkylierung der Cysteine geprüft werden. MEL wurde mit TCEP-reduziertem OT im NaH2PO4-Puffer bei pH 8.5 im molaren Verhältnis 4:3 umgesetzt. Die Kontrollreaktion wurde ohne MEL durchgeführt. Zu diesen Lösungen wurde Iodacetamid (IAA) im molaren Verhältnis MEL / OT / IAA 4:3:130 zugegeben. Nach 30 min Reaktionszeit wurde weiterhin DTT zugegeben, so daß ein molares Verhältnis MEL / OT / IAA / DTT von 4:3:130:50 resultierte. UV-spektroskopischen Untersuchungen sind aufgrund der großen Eigenabsorption von Iodacetamid (bzw. der Hydrolyseprodukte) im Bereich von 220-280 nm nicht möglich.
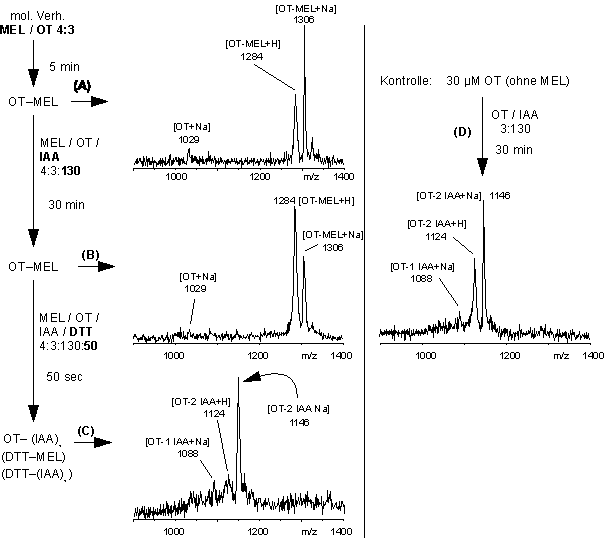
Abb. 63: MEL als Schutzgruppe für räumlich nahe Biscysteinylfunktionen in Vasopressin und die anschließende Alkylierung der Cysteine. Die Sequenz der MALDI-Massenspektren zeigt (A) OT-MEL im NaH2PO4 -Puffer bei pH 8.5 vor und (B) 30 min nach Zugabe von IAA im molaren Verhältnis MEL / OT / IAA 4:3:130, (C) 50 Sekunden nach Zugabe von DTT im molaren Verhältnis MEL / OT / IAA / DTT 4:3:130:50.
Aliquote der Reaktionslösungen wurden MALDI-massenspektrometrisch untersucht. Die MALDI-Massenspektren (Abb. 63 A und B) vor und 30 min nach Zugabe von IAA zeigen nur MEL-modifiziertes OT (m/z 1284) bzw. das Na-Addukt (m/z 1306). 50 sec. nach Zugabe von DTT (Abb. 63 C) liegt OT hauptsächlich zweifach carboxamidomethyliertes OT (OT-(IAA)2, m/z 1124, Na-Addukt m/z 1146) vor. Geringe Mengen an einfach alkyliertem OT-IAA (Na-Addukt 1088) sind nach 3 h vollständig zu OT-(IAA)2 abreagiert (nicht gezeigt). Im Vergleich zu VP (Abb. 61) erfolgen keine Dreifachmodifizierungen, wie die Kontrolle nach 30 min Reaktionsdauer zeigt (Abb. 63 D). Bemerkenswert ist die quantitative Spaltung des OT-MEL-Addukts durch DTT, obwohl nur ein 1.2-facher molarer Überschuß an IAA bezogen auf die Gesamtthiolkonzentration vorlag. Die Alkylierung der Cysteine kann auf eine Umesterungsreaktion von Vas-MEL (durch DTT zu Vas und DTT-MEL) und anschließende Reaktion der Cysteinylthiolgruppen mit IAA zurückgeführt werden. Als Nebenreaktion wäre die Reduktion einer eventuell ausgebildeten Disulfidbrücke durch DTT denkbar.
Damit kann MEL als tertiärstrukurselektive Schutzgruppe für räumlich nahe Cysteinreste in Proteinen eingesetzt werden. InAbb. 64 ist das hier vorgeschlagene analytische Konzept zur Identifizierung räumlicher naher Cysteinreste verdeutlicht. Cysteine, die nicht durch MEL geschützt werden, weil sie nur offenkettige und damit instabilere Arsonigsäuremono- oder dithioester bilden, können durch ein cysteinselektives Reagenz modifiziert werden. Nach Abtrennung der überschüssigen Reagenzien wird in Gegenwart eines zweiten cysteinselektiven Modifizierungsreagenz die MEL-Schutzgruppe mit einem Dithiol abgespalten und damit gleichzeitig die entschützten Cysteine modifiziert.
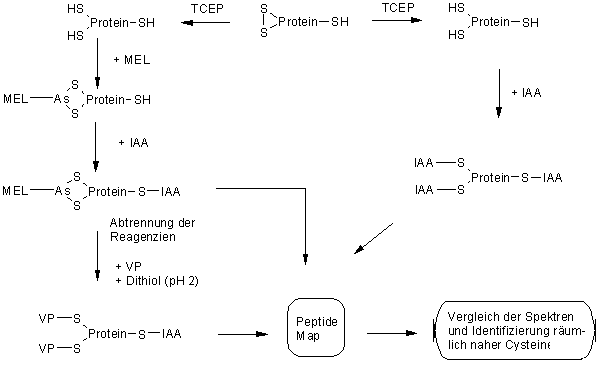
Abb. 64: Vorgeschlagenes analytisches Konzept zum Einsatz von MEL als tertiärstrukurselektive Schutzgruppe für räumlich nahe Cysteinreste in Proteinen.
Die denkbare Reduktion partiell ausgebildeter Disulfidstrukturen, z.B. von Faltungsintermediaten, durch das Dithiol könnte dadurch verhindert werden, daß die Abspaltungsreaktion von MEL im sauren pH-Bereich durchgeführt wird (siehe 2.4.3). Durch chemische und proteolytische Abbaumethoden (siehe 2.6.3) können dann die einzelnen Cysteinmodifizierungen analysiert und anhand des modifizierten Rests entweder einem freien oder mit MEL geschützten Cystein zugeordnet werden. Weitere experimentelle Untersuchungen wurden im Rahmen dieser Arbeit nicht durchgeführt.
|
|
|
|
|
2.6 Selektive chemische Modifizierung von Modellproteinen mit Melarsenoxid und massenspektrometrische Charakterisierung der Reaktionsprodukte |
|
|
2.4 Untersuchungen zur Stabilität der Reaktionsprodukte von mit Melarsen Oxid modifizierten Modellpeptiden |
Diese Seite wurde erstellt am 15. März 1998. Letzte Änderungen am 30. Juni 1998 durchPeter Happersberger.