|
|
|
|
|
2.4 Untersuchungen zur Stabilität der Reaktionsprodukte von mit Melarsen Oxid modifizierten Modellpeptiden |
|
|
2.2 UV-spektroskopische Untersuchungen zum Reaktionsmechanismus der Modifizierung von Bis-Thiolen mit Melarsen Oxid |
2.3 Massenspektrometrische Charakterisierung der Reaktionsprodukte von Modellpeptiden mit Arsonigen Säuren
Zur massenspektrometrischen Untersuchung der biscysteinylthiolselektiven Modifizierung mit Derivaten der Arsonigen Säure wurde eine Auswahl an Modellpeptiden getroffen, die den folgenden Ansprüchen genügen:
- Die Peptide sollten nur zwei Thiolfunktionen entweder als freie Cysteine oder als Disulfidbrücke besitzen, die mit den Arsonigen Säuren zu zyklischen Arsonigsäuredithioestern reagieren. Die Selektivität gegenüber Biscysteinylfunktionen und mögliche Nebenreaktionen mit anderen Aminosäuren sollten somit ermittelbar sein.
- Das Molekulargewicht der zu untersuchenden Peptide sollte im Bereich von 0.5 bis 5 kD liegen, so daß auch bei der Anwendung massenspektrometrischer Methoden mit geringer Auflösung die Differenzierung und Identifizierung der entstehenden Umsetzungsprodukte eindeutig ist.
- Wenn möglich, sollten proteolytische Spaltstellen zur Verfügung stehen, um über proteolytische Abbaupeptide die Art und Position der Modifizierung zu bestimmen. Außerdem sollten Informationen über die Stabilität der Modifizierung unter den Abbaubedingungen gewonnen werden.
Folgende Peptide wurden für die massenspektrometrische Analyse für die Umsetzung mit den Arsonigen Säuren verwendet.
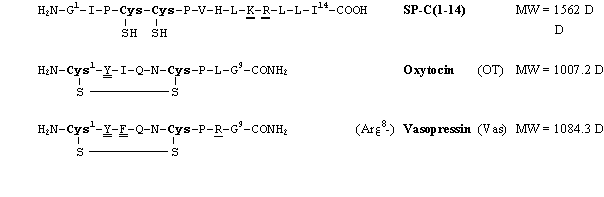
Abb. 43: Aminosäuresequenzen und Molekulargewichte von Peptiden zur massenspektrometrischen Charakterisierung der Umsetzung mit Arsonigen Säuren. Gezeigt sind zusätzlich die N- und C-Termini. In fetter Schrift sind die Aminosäuren Cystein gezeigt. Unterstrichen sind tryptische, doppelt unterstrichen sind -chymotryptische Spaltstellen.
Vasopressin ist wie Oxytocin (vgl. 2.2.1) ein zyklisches - über eine Disulfidbrücke stabilisiertes -Peptid aus neun Aminosäuren. Bis auf die Aminosäuren drei und acht sind die Sequenzen identisch (sieheAbb. 43). In beiden Peptiden sind die Cysteine durch vier weitere Aminosäuren getrennt. Oxytocin wurde v.a. zur UV-spektroskopischen Charakterisierung der Modifizierungsreaktion (siehe 2.2) eingesetzt, während Vasopressin aufgrund seiner tryptischen und -chymotryptischen Spaltstellen für Stabilitätsuntersuchungen verwendet wurde.
Die Sequenz des synthetischen Peptids SP-C(1-14) entspricht den ersten 14 Aminosäuren des Lungensurfactant-Proteins-C (siehe 2.6.1). Es enthält zwei vicinale Cysteinylreste, die reduziert vorliegen.
2.3.1 Umsetzung von Dicysteinylpeptiden mit Derivaten der Arsonigen Säure
Im Peptid SP-C(1-14), dem einfachsten Modellsystem, liegen die beiden Cysteine vicinal in reduzierter Form vor, so daß eine Reduktion nicht notwendig ist. Synthetisches SP-C(1-14) stand in lyophilisierter Form zur Verfügung. Zum Zeitpunkt dieser Versuche lagen die Ergebnisse - hinsichtlich Stöchiometrie und Selektivität - aus den UV-spektroskopischen Untersuchungen (siehe 2.2) noch nicht vor. Die jeweiligen Arsonigsäurederivate PAA, PYR, A-PAA und MEL wurden als konzentrierte Stammlösungen in organischen Lösungsmittel(gemischen) eingesetzt (siehe 3.4.3). Die Lösung von A-PAA war bereits eine Woche gelagert (siehe 2.1.4), die der anderen Derivate waren frisch zubereitet. Um Modifizierungsbedingungen zu gewährleisten, wie sie für die Proteinfaltungsreaktionen als notwendig erachtet wurden, sollte der Anteil an organischem Lösungsmittel 5% (v/v) (Endkonzentration) nicht übersteigen. Die Modifizierung wurde in NH4OAc-Puffer bei pH 4.0 durchgeführt, da SP-C bei höheren pH-Werten zu nicht spezifischer Agglomeration [135]. Das molare Verhältnis von SP-C(1-14) zu Arsoniger Säure betrug für PAA 1:4.3, für MEL, PYR und A-PAA jeweils 1:23 (siehe 3.4.3). Anschließend wurden die überschüssigen Reagenzien über SepPak C18-RP Kartuschen (siehe 3.6.1) entfernt. Die Abtrennung von PAA, PYR und A-PAA gelingt vollständig, während das deutlich hydrophobere MEL (vgl. HPLC-Reinigung, S. 53) z.T. zusammen mit dem Peptid eluiert. Die so gereinigten Peptide wurden nach Lyophilisieren mittels MALDI-MS charakterisiert.
Das MALDI-Massenspektrum des unmodifizierten SP-C(1-14) (sieheAbb. 44 A) zeigt neben dem protonierten Molekülion [SP-C(1-14)+H]+ bei m/z 1563 Natrium-, Kaliumaddukte (m/z 1565, 1601) und die für reduziert vorliegende Peptide typischen HCCA-Addukte (m/z 1752). Die Modifizierungen mit PAA, PYR und MEL (Abb. 44 C-E) erfolgen fast quantitativ zum einfach modifizierten SP-C(1-14)-PAA (m/z 1713), SP-C(1-14)-PYR (m/z 1714) und SP-C(1-14)-MEL (m/z 1837). Es sind keine unspezifischen Mehrfachmodifizierungen zu erkennen. Das MALDI-Massenspektrum für die Modifizierung mit A-PAA (Abb. 44 B) zeigt uneinheitliche Reaktionsprodukte. Neben nicht modifizierten (m/z 1563) und einfach modifizierten SP-C(1-14)-A-PAA (m/z 1728) erkennt man Signale bei m/z 1824 und 2081. Die Massendifferenzen m = 261 und 351, zum nicht modifizierten SP-C(1-14), entsprechen Peaks im ESI-Massenspektrum von "gealtertem" A-PAA (siehe Abb. 32). Die Signale bei m/z 1824 und 2081 könnten somit Modifizierungen durch "Alterungsprodukte" von A-PAA entsprechen.
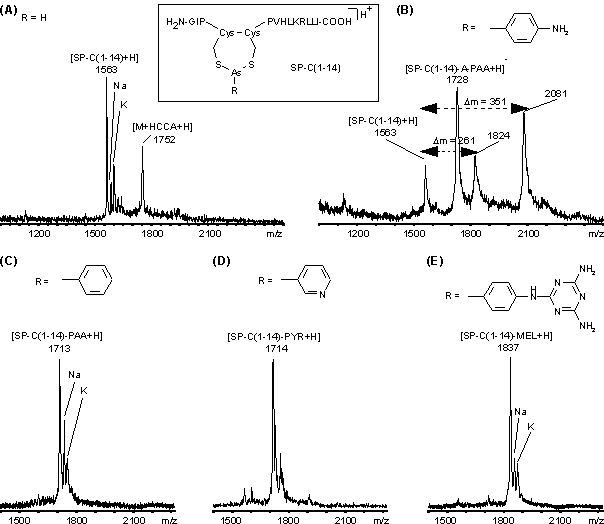
Abb. 44: MALDI-Massenspektren von (A) nicht modifiziertem SP-C(1-14) und der Eluate, der über C18-RP-SepPak-Kartuschen gereinigten Reaktionslösungen der Umsetzung von SP-C(1-14) mit (B) A-PAA, (C) PAA, (D) PYR und (E) MEL.
2.3.2 Umsetzung von cystinylhaltigen Modellpeptiden mit Derivaten der Arsonigen Säure
Reaktion von nicht-reduziertem Oxytocin mit Melarsen Oxid als Kontrollexperiment für die Rückfaltung und anschließende in situ-Reduktion
Um die chemische Selektivität der Arsonigen Säuren für Cysteinylfunktionen im Vergleich zu Cystinylfunktionen zu überprüfen wurden 45 µM Melarsen Oxid (MEL) mit 30 µM nicht reduziertem Oxytocin (OT) bei pH 4 und 8 versetzt (siehe 3.4.3) und die Reaktion UV-spektroskopisch verfolgt (siehe 2.2). Die Änderung der Absorptionsdifferenz mit der Zeit ![]() OD299-267 (t) ist in Abb. 45 A dargestellt. Sowohl bei pH 8.2 (120 s t 600 s) als auch bei pH 4 (120 s t 900 s) reagiert MEL nicht mit nicht reduziertem OT:
OD299-267 (t) ist in Abb. 45 A dargestellt. Sowohl bei pH 8.2 (120 s t 600 s) als auch bei pH 4 (120 s t 900 s) reagiert MEL nicht mit nicht reduziertem OT: ![]() OD299-267(t) bleibt konstant. Erst nach Zugabe von 150 µM TCEP zur Reaktionslösung steigt
OD299-267(t) bleibt konstant. Erst nach Zugabe von 150 µM TCEP zur Reaktionslösung steigt ![]() OD299-267 (t). Vergleicht man die Zeitdauer zum Erreichen des Plateaus von OD299-267(t) für die Reaktion von MEL mit bereits reduziertem OT (S. 42) erkennt man, daß der geschwindigkeitsbestimmende Schritt nicht die Reaktion von MEL mit reduziertem OT, sondern die Reduktion von OT durch TCEP ist. Die Kurven inAbb. 45 A geben also direkt die Reduktionsgeschwindigkeiten von TCEP für verschiedene pH-Werte bei RT wieder. TCEP reduziert OT bei pH 8.2 innerhalb von 300 s vollständig, während die Reduktion bei pH 4 selbst nach Zugabe weiterer 500 µM TCEP mehr als 90 min dauert. Dies stimmt mit der Literatur [134] überein. Das Absinken der Kurve
OD299-267 (t). Vergleicht man die Zeitdauer zum Erreichen des Plateaus von OD299-267(t) für die Reaktion von MEL mit bereits reduziertem OT (S. 42) erkennt man, daß der geschwindigkeitsbestimmende Schritt nicht die Reaktion von MEL mit reduziertem OT, sondern die Reduktion von OT durch TCEP ist. Die Kurven inAbb. 45 A geben also direkt die Reduktionsgeschwindigkeiten von TCEP für verschiedene pH-Werte bei RT wieder. TCEP reduziert OT bei pH 8.2 innerhalb von 300 s vollständig, während die Reduktion bei pH 4 selbst nach Zugabe weiterer 500 µM TCEP mehr als 90 min dauert. Dies stimmt mit der Literatur [134] überein. Das Absinken der Kurve ![]() OD299-267 (t) bei pH 8.2 nach ca. 1500 s läßt auf eine partielle Reoxidation des Oxytocins schließen. Nach erneuter Zugabe von TCEP (t 3300 s) erfolgt ein leichter Anstieg von
OD299-267 (t) bei pH 8.2 nach ca. 1500 s läßt auf eine partielle Reoxidation des Oxytocins schließen. Nach erneuter Zugabe von TCEP (t 3300 s) erfolgt ein leichter Anstieg von ![]() OD299-267 (t). Offensichtlich ist das OT-MEL-Addukt unter diesen Bedingungen deutlich weniger stabil als erwartet, so daß die Rückbildung der Disulfidbrücke im basischen Mileau erfolgt. Die erneute Reduktion durch TCEP erfolgt solange, bis der Überschuß an TCEP aufgebraucht ist. Erst dann nimmt
OD299-267 (t). Offensichtlich ist das OT-MEL-Addukt unter diesen Bedingungen deutlich weniger stabil als erwartet, so daß die Rückbildung der Disulfidbrücke im basischen Mileau erfolgt. Die erneute Reduktion durch TCEP erfolgt solange, bis der Überschuß an TCEP aufgebraucht ist. Erst dann nimmt ![]() OD299-267 (t) mit der Zeit langsam ab. Auf die Stabilität der Produkte in verschiedenen Puffersystemen wird in Abschnitt 2.4.2 eingegangen. Da selbst nach Zugabe von hohen molaren Überschüssen an TCEP (t 3600 s für pH 8.2 bzw. t 6300 s für pH 4)
OD299-267 (t) mit der Zeit langsam ab. Auf die Stabilität der Produkte in verschiedenen Puffersystemen wird in Abschnitt 2.4.2 eingegangen. Da selbst nach Zugabe von hohen molaren Überschüssen an TCEP (t 3600 s für pH 8.2 bzw. t 6300 s für pH 4) ![]() OD299-267 (t) konstant bleibt, war die Reduktion zu diesen Zeitpunkten abgeschlossen. Das MALDI-Massenspektrum (Abb. 45 B) für den Zeitpunkt t 600 s (vor der Zugabe von TCEP) zeigt nicht modifiziertes Oxytocin bei m/z 1008 bzw. für t 6000 s nahezu vollständig modifiziertes Oxytocin (OT-MEL: m/z 1283). Die MALDI-Massenspektren sind für beide pH-Werte identisch.
OD299-267 (t) konstant bleibt, war die Reduktion zu diesen Zeitpunkten abgeschlossen. Das MALDI-Massenspektrum (Abb. 45 B) für den Zeitpunkt t 600 s (vor der Zugabe von TCEP) zeigt nicht modifiziertes Oxytocin bei m/z 1008 bzw. für t 6000 s nahezu vollständig modifiziertes Oxytocin (OT-MEL: m/z 1283). Die MALDI-Massenspektren sind für beide pH-Werte identisch.
Melarsen Oxid reagiert also nicht mit intakten Disulfidbrücken, was eine wichtige Voraussetzung für das Abfangen von Faltungsintermediaten ist (siehe 0).

Abb. 45: (A) Änderung der Absorptionsdifferenz OD299-267 (t) für die Reaktion von MEL mit (nicht reduziertem) Oxytocin (OT) bei pH 8.2 und pH 4.0. Erst nach Zugabe von TCEP erfolgt die Reduktion von OT und die Reaktion mit MEL zum Arsonigsäuredithioester.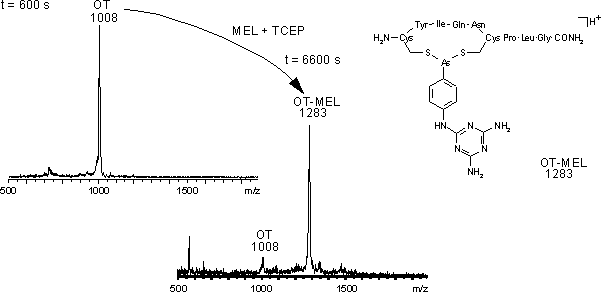
Abb. 46: MALDI-Massenspektren für die Zeitpunkte t 600 s (vor TCEP-Zugabe) und t 6600 s ausAbb. 45. Die MALDI-Massenspektren sind für pH 4 und pH 8 identisch.
Umsetzung von partiell reduziertem Vasopressin mit dem Anhydrid der Pyridin-3-arsonigen Säure (PYR)
Der zeitliche Verlauf der Reduktion von Vasopressin mit DTT im NH4HCO3-Puffer bei pH 8.3 wurde FAB-massenspektrometrisch verfolgt (sieheAbb. 47 A und B). Beim Verhältnis an oxidiertem zu reduziertem Vasopressin von ca. 1:2 wurde die Reduktion durch 10-faches Verdünnen mit NH4OAc-Puffer (pH 3.5) gestoppt. Die Modifizierung mit PYR erfolgte im molaren Verhältnis von Vasopressin / DTT / PYR 1:5:6 (siehe 3.4.3). Das MALDI-Massenspektrum (nicht dargestellt) der Reaktionslösung nach Lyophilisieren zeigte nicht modifiziertes Vasopressin zu Vas-PYR im Verhältnis von 1:2. Unspezifische Zweitmodifizierungen waren nicht vorhanden. Das nach C18-RP-HPLC-Reinigung von den lyophilisierten HPLC-Fraktionen erhaltene ESI-Massenspektrum (sieheAbb. 47 C) zeigte nahezu identische Intensitätsverhältnisse von ein- bzw. zweifach protoniertem Vasopressin (m/z 1085, 544) und Vas-PYR (m/z 1238, 620). Des weiteren sind sequenz-unspezifische Fragmentionen zu erkennen. Die bei der Firma Bruker-Franzen aufgenommenen hochaufgelösten ESI-FT-ICR-Massenspektren (Electrospray- Ionization- Fourier- Transformation- Ion-Centrocon- Resonance- Mass-Spectrometry) sollten zeigen, ob das gesamte reduziert vorliegende Vasopressin mit PYR reagiert hatte. Im Falle einer nur teilweise erfolgten Modifizierung sollte reduziertes neben oxidiertem Vasopressin vorliegen (m = 2). Die ESI-FT-ICR-Massenspektren wurden mit einer Massenauflösung von 30000 aufgenommen und ermöglichen so eine genaue Massenbestimmung auf die vierte Nachkommastelle. Probenpräparationsbedingt wurden nur die zweifach protonierten Molekülionen beobachtet. Die inAbb. 47 D dargestellten Ausschnitte der ESI-FT-ICR-Massenspektren der HPLC-Fraktion von Vas-PYR belegen eindeutig den oxidierten Charakter des nicht modifizierten Vasopressins. Reduziert, nicht modifiziert vorliegendes Vasopressin konnte nicht nachgewiesen werden. Die erhaltenen Intensitäten stimmen sehr gut mit den theoretisch berechneten Isotopenverteilungen (Inserts in Abb. 47 D) überein. Diese Daten belegen eindeutig - in Übereinstimmung mit den UV-spektroskopischen Untersuchungen an Oxytocin (s.o.) - die chemische Selektivität der Arsonigen Säuren für Biscysteinylgruppierungen und das indifferente Verhalten gegenüber Disulfidstrukturen in Proteinen.
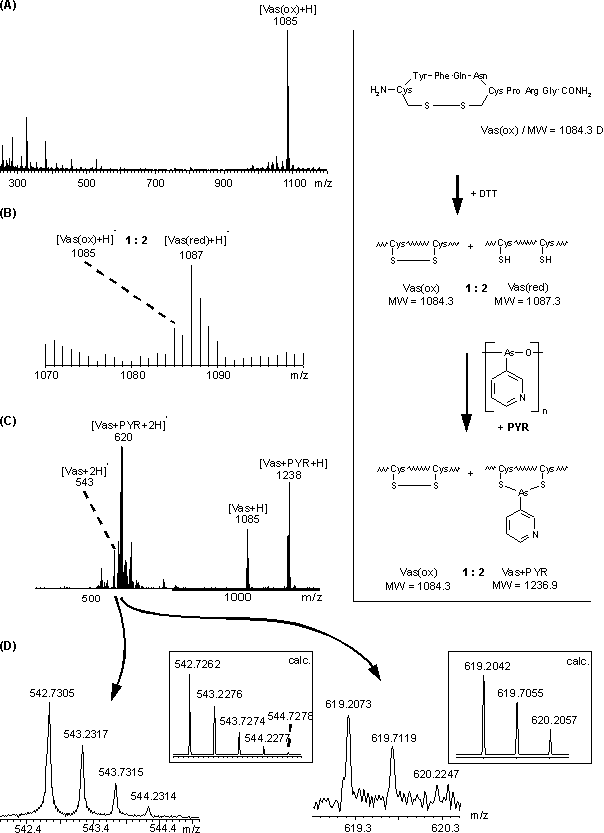
Abb. 47: (A) FAB-Massenspektrum von nicht reduziertem Vasopressin, (B) FAB-Massenspektrum von partiell reduziertem Vasopressin (oxidiert / reduziert 1:2) (C) ESI-Massenspektrum des mit PYR umgesetzten partiell reduzierten Vasopressins. (D) Hochaufgelöste ESI-FT-ICR-Massenspektren und berechnete Isotopenverteilungen der zweifach protonierten Molekülionen der gleichen Reaktionslösung aus (C).
Quantitative Umsetzung von vollständig reduziertem Vasopressin mit Derivaten der Arsonigen Säure
Die Reaktionsprodukte aus Vasopressin (Vas) und den synthetisierten Arsonigen Säuren PAA, MEL, PYR und A-PAA sollten isoliert und massenspektrometrisch charakterisiert werden. Dazu wurde Vasopressin (a) im NH4HCO3-Puffer bei pH 8.0 mit DTT und (b) im NH4OAc-Puffer bei pH 4.0 mit TCEP vollständig reduziert (siehe 3.4.1). Die Modifizierungsreaktion erfolgte in einem molaren Verhältnis Vasopressin / Reduktionsmittel / Arsonige Säure = 1:5.4:13 bei pH 4 (TCEP-Reduktion) bzw. pH 8 (DTT-Reduktion). Berücksichtigt man die stöchiometrische Reaktion der Arsonigen Säuren mit DTT resultiert ein molares Verhältnis Vasopressin / Arsoniger Säure = 1:7.6 (siehe 3.4.3). Die Arsonigsäurederivate wurden als konzentrierte Stammlösungen in organischen Lösungsmittelgemischen eingesetzt. Der Anteil an organischem Lösungsmittel während der Modifizierung betrug für PAA 55% (v/v) DMSO bzw. 4% (v/v) MeOH für die restlichen Derivate. Die aus der Reaktionslösung ausgefallenen Niederschläge wurden abzentrifugiert, die Überstände abgetrennt. Bei den Niederschlägen handelt es sich vermutlich um die Reaktionsprodukte aus den Arsonigsäure-Derivaten und DTT bzw. um die überschüssigen Arsonigsäure-Derivate, die in wäßrigem Mileau deutlich schlechter löslich sind. Die MALDI-Massenspektren der Überstände für die Modifizierungsreaktion bei pH 8 zeigt Abb. 49; in Abb. 48 sind die entsprechenden Strukturen und berechneten m/z-Werte gezeigt.
Die Spektren zeigen für pH 8 unterschiedliche Anteile an umgesetzten Vasopressin. PAA reagiert praktisch vollständig zu Vas-PAA (m/z 1237) (Abb. 49 A), während für MEL nur zu einem geringen Anteil Vas-MEL (m/z 1361) vorliegt (Abb. 49 C). Schätzt man die Quantität der Umsetzung über die relativen Intensitäten ab, liegt Vasopressin für PYR und A-PAA je zu etwa 40% als Vas-PYR (m/z 1238) und Vas-A-PAA (1251) vor. (Abb. 49 B und D). Anzumerken ist noch, daß MALDI-Massenspektren von Peptiden (oder Proteinen), die mit den hier verwendeten Derivaten der Arsonigen Säure modifiziert wurden, typische Matrixaddukte zeigen (siehe 2.4.1). Als Matrix für die MALDI-MS-Untersuchungen wurde HCCA verwendet; die entsprechenden Signale sind in Abb. 49 mit "HCCA" abgekürzt. Die unterschiedlichen Modifizierungsgrade bei pH 8 überraschten zunächst, da die UV-spektroskopischen Untersuchungen (siehe 2.2.2) gezeigt hatten, daß Umsetzungen von MEL mit Dithiolen bei pH 8 quantitativ und schnell erfolgen. Vermutlich erfolgte die Modifizierung von Vasopressin bei pH 8 auch quantitativ, wie die vollständige Modifizierung von Vasopressin mit PAA zu Vas-PAA zeigt. Der Grund für die unterschiedlichen Modifizierungsgrade war das längere Stehenlassen der Reaktionsüberstände bei Raumtemperatur (3-5 h). Wie in Abschnitt 2.4.2 gezeigt, hydrolysiert zumindestens Vas-MEL nach Abtrennung der Reaktionsüberschüsse (s.u.) in 50 mM NH4HCO3 pH 8 innerhalb einer Stunde praktisch quantitativ zum unmodifizierten Peptid. So sind die unterschiedlichen Modifizierungsgrade dadurch zu erklären, daß die Vassopressin- Arsonigsäure- Derivate z.T. hydrolysierten. MEL, das in wäßrigen Puffern am geringsten lösliche Derivat fällt dabei aus (Beleg: Niederschlag im Überstand) und verschiebt somit das Hydrolysegleichgewicht vollständig auf die Seite von unmodifiziertem Vasopressin (Abb. 49 C).
Für PYR und A-PAA, die in Wasser besser löslich sind, erhält man noch zum Teil modifiziertes Peptid (Abb. 49 B und D). PAA bleibt wahrscheinlich bei einen organischen Lösungsmittelanteil von 55% (v/v) DMSO vollständig in Lösung. Auch geringen Mengen an freiem DTT, durch partielle Hydrolyse des entsprechenden Arsonigsäuredithioesters, kann wie in 2.4.3 gezeigt wird, die Spaltung der As-S-Peptidbindung bewirken.
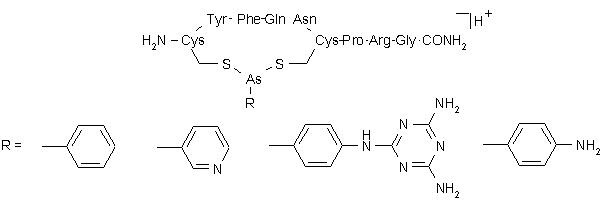
Abb. 48: Vorgeschlagende Strukturen und berechnete m/z-Werte für MH+ für die Reaktion von reduziertem Vasopressin mit PAA, PYR, MEL und A-PAA.
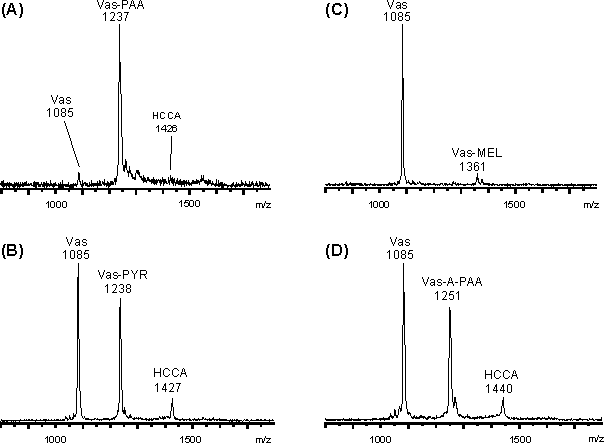
Abb. 49: MALDI-Massenspektren der Überstände für die Reaktion von reduziertem Vasopressin mit (A) PAA, (B) PYR, (C) MEL und (D) A-PAA bei pH 8. Die mit "HCCA" gekennzeichneten Signale stellen Addukte aus MALDI-Matrix und modifizierten Vasopressin dar.
Die MALDI-Massenspektren der Überstände für die Modifizierung bei pH 4 sind in Abb. 50 gezeigt. PAA und MEL reagieren praktisch quantitativ zu Vas-PAA (m/z 1237) und Vas-MEL (m/z 1361) (Abb. 50 A und C). Für A-PAA liegt Vasopressin zu 77% als Vas-A-PAA (m/z 1251) und für PYR zu 68% als Vas-PYR (m/z 1238) vor (Abb. 50 D und B). Die durchweg höheren Umsetzungsgrade bei pH 4 stehen im Einklang mit der größeren Stabilität von Vas-MEL bei pH 4 (siehe 2.4.2). Das Spektrum für PYR (Abb. 49 B) zeigt unspezifische Zweifach-Modifizierungen bei m/z 1389. Die Massendifferenz m = 151 zum einfach modifizierten Vas-PYR zeigt eine kovalente Bindung des zweiten PYR-Moleküls an das Peptid. Denkbare weitere Modifizierungsstellen am Peptid wären der N-Terminus und die Aminosäuren Tyrosin und Arginin. Eventuell spielt auch der oligomere Charakter von PYR für die Zweitmodifizierung eine Rolle (vgl. Abb. 26). Die für die Addukte aus MALDI-Matrix (HCCA) und arsonigsäurederivatisiertem Vasopressin erhaltenen Signale sind in Abb. 50 als "HCCA" abgekürzt.
Die Reaktionsgemische Vas-PAA, Vas-PYR, Vas-MEL und Vas-A-PAA der Umsetzung bei pH 4 wurden über eine C18-RP-HPLC getrennt und die erhaltenen Produkte über MALDI-, PD- und ESI-MS charakterisiert.Abb. 51 A zeigt das HPLC-Chromatogramm der Umsetzung von Vasopressin mit MEL zu Vas-MEL bei pH 4.
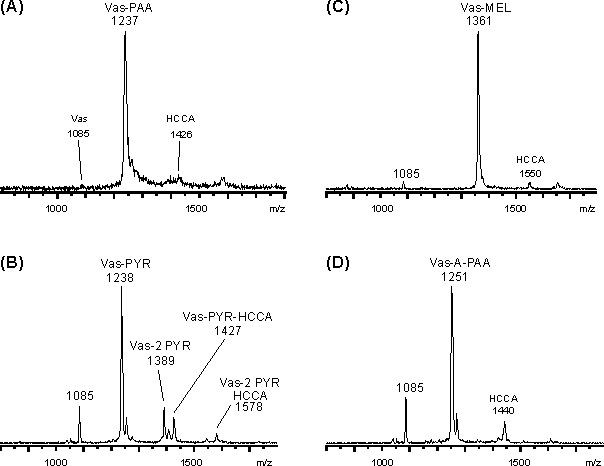
Abb. 50: MALDI-Massenspektren der Überstände für die Reaktion von reduziertem Vasopressin (A) PAA, (B) PYR, (C) MEL und (D) A-PAA bei pH 4. Die mit "HCCA" gekennzeichneten Signale stellen Addukte aus MALDI-Matrix und modifizierten Vasopressin dar.
Der Vergleich der Retentionszeiten der HPLC-Analysen zeigt, daß die Umsetzung nahezu quantitativ zum einfach-modifizierten Vas-MEL erfolgte. Aufgrund der Modifizierung mit MEL ist das Vas-MEL deutlich hydrophober als Vas und eluiert bei höheren Retentionszeiten (LegendeAbb. 51 A). Die nicht basisliniengetrennten Peaks 3 und 4 entsprechen vermutlich den beiden diastereomeren Arsonigsäuredithioestern (siehe Abb. 8), da die experimentell bestimmten Molekulargewichte nach MALDI-MS identisch sind. Das MALDI-Massenspektrum (Abb. 51 B) der nicht lyophilisierten HPLC-Fraktion 3 zeigt neben dem modifizierten Peptid Vas-MEL (m/z 1361) in geringen Anteilen unmodifiziertes Vasopressin (m/z 1085) und das HCCA-Matrix-Addukt von Vas-MEL (1550). Da die Fraktionen nach HPLC-Trennung im sauren Mileau (pH 2) vorliegen stellt sich vermutlich das säurekatalysierte Gleichgewicht der Veresterungsreaktion zwischen MEL, Vasopressin und Vas-MEL erneut ein und erklärt den gefundenen Anteil an unmodifiziertem Vasopressin nach HPLC-Reinigung. Dies bestätigt das PD-Massenspektrum (Abb. 51 C) der gleichen Probe nach Lyophilisieren. Man erhält praktisch identische Intensitätsverhältnisse wie bei der MALDI-MS-Analyse. Die HPLC-Chromatogramme, MALDI- und PD-Massenspektren für Vas-PAA und Vas-A-PAA zeigen analoge Ergebnisse zu Vas-MEL.
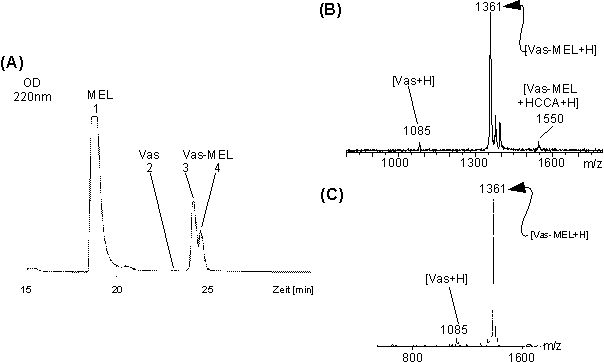
Abb. 51: (A) HPLC-Chromatogramm der Umsetzung von Vasopressin mit MEL zu Vas-MEL. Das Chromatogramm zeigt neben den beiden Produktpeaks (vermutlich Diastereomere) bei t = 23.9 (Fr. 3) und 24.3 min (Fr. 4) noch Signale für überschüssiges MEL (t = 18.4 min, Fr. 1) und nicht modifiziertes Vasopressin (t = 22.4, Fr. 2). Die Fraktionen wurden über MALDI- und PD-MS charakterisiert. Das MALDI-MS (B) und PD-MS (C) von Fr. 3 zeigt die nahezu quantitative Umsetzung zu Vas-MEL. Die Spektren sind für beide HPLC-Fraktionen 3 und 4 identisch.
Die C18-RP-HPLC-Trennung der Reaktionsprodukte aus der Umsetzung von Vasopressin mit PYR erfolgt dagegen deutlich schlechter.Abb. 52 D zeigt das HPLC-Chromatogramm. Aus dem Vergleich der Retentionszeiten kann nicht auf den Grad der Umsetzung geschlossen werden, da nicht modifiziertes Vasopressin mit Peak 1 und 2 gefunden wurde. Die gesammelten Fraktionen wurden über MALDI-MS und PD-MS charakterisiert. Die MALDI- und PD-Massenspektren von Fr. 1 (Abb. 52 A und B) zeigen Signale für Vasopressin (m/z 1085), Vas-PYR (m/z 1238) und von zweifach modifiziertem Vasopressin (m/z 1389); während das MALDI-Massenspektrum von Fr. 2 (Abb. 52 C) neben Vas (m/z 1085) nahezu ausschließlich einfach modifiziertes Vas-PYR (m/z 1238) zeigt. Weiterhin lassen sich Signale den Addukten aus MALDI-Matrix (HCCA) und ein- und zweifach modifizierten Vasopressin (m/z 1427 und 1578) zuordnen. Die HPLC-Fraktionen 1 und 2 enthalten im Vergleich zu Vas-MEL (Abb. 51 B) und zum Reaktionsüberstand vor der HPLC-Reinigung (Abb. 50 B) deutlich höhere Anteile an unmodifizierten Vasopressin. Nach Abtrennung der Reaktionsüberschüsse stellt sich das säurekatalysierten Gleichgewicht von Vas, PYR und Vas-PYR erneut ein (s.o.). Im Vergleich zu Vas-MEL liegt das Gleichgewicht weiter auf der Seite der Edukte, was auf eine geringeren Stabilität von Vas-PYR schließen läßt. Vor allem wegen der beobachteten unspezifischen Zweifachmodifizierung eignet sich PYR nicht als biscysteinylselektives Modifizierungsreagenz.
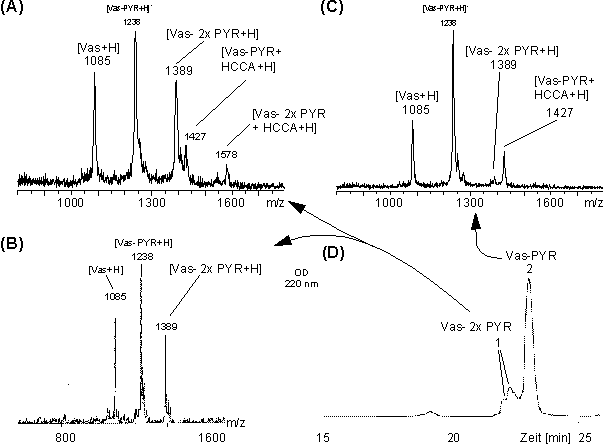
Abb. 52: Das (A) MALDI- und (B) PD-Massenspektrum der HPLC-Fraktion 1 (t = 21.9 min) zeigt nicht, einfach und zweifach modifiziertes Vasopressin. Das MALDI-Massenspektrum (C) von Fraktion 2 (t = 22.7 min) zeigt nahezu nur einfach modifiziertes Vas-PYR. Mit HCCA sind die HCCA-Addukte bezeichnet. (D) Dargestellt ist das zugehörige HPLC-Chromatogramm der Trennung von Vas-PYR.
Die ESI-MS Analyse von Vas-MEL wurde bei verschiedenen Repellerspannungen (5 - 80 V) durchgeführt. Durch höhere Repellerspannungen (> 20 V) lassen sich Fragmentierungen des Probenmoleküls einleiten. Die Molekülionen erfahren eine zusätzliche Beschleunigung bevor sie in den Analysator eintreten. Abhängig von Druck, Temperatur und Verweildauer der Ionen nimmt die Zahl und die durchschnittliche Energie der Stöße mit restlichen Gasmolekülen im Massenspektrometer zu. Analog begründet sich die Verschiebung der Ladungsverteilung zu niedriger geladenen Molekülionen. Mit zunehmender Repellerspannung "fragmentieren" die nicht kovalent gebunden Protonen zunehmend vom Analytmolekül. Dies kann für sehr hohe Repellerspannungen (> 80 V) zur vollständigen Unterdrückung von Molekülionen, und damit zu niedrigen Signalintensitäten und hohen Rauschanteilen der Spektren führen. Diese sog. "Spikes" treten bei jedem Frequenzdurchgang des Quadrupols bei anderen m/z-Werten auf, sie sind somit irreproduzierbar und entsprechen keiner Isotopenverteilung. Nach M. H. Allen und M. Vestal defokussieren hohe Repellerspannungen den Ionenstrahl, was zu einer geringeren Transmission der Ionen durch die Quelle in den Massenanalysator führt [136].
Die Spaltung an der C-CO Bindung, der Peptidbindung CO-NH oder der NH-C Bindung einer Peptidkette führt zu sequenzspezifischen Fragmenten. Außerdem sind Wasserabspaltungen und Fragmentierungen der Seitenketten möglich. Nach der von Roepstorff und Fohlmann vorgeschlagenen Nomenklatur [137] werden bei Ladungserhaltung am N-Terminus, die N-terminalen Fragmentionen mit A, B und C bei Ladungserhaltung am C-Terminus, die C-terminalen Fragmentionen mit X,Y und Z bezeichnet. Eine Indexzahl bezeichnet jeweils die Nummer des Aminosäurerestes nach dem abgespalten wird, wobei die N-terminalen Fragmentionen vom N-Terminus, die C-terminalen ab dem C-Terminus bezeichnet werden. Umlagerungen, die zu zusätzlichen Protonierungen führen, werden durch hochgestellte Striche gekennzeichnet (sieheAbb. 53).
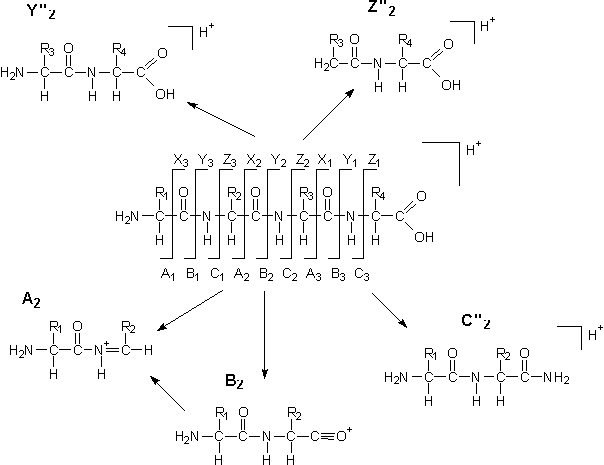
Abb. 53: Fragmentierungsschema für Peptide, mit der nach Roepstorff und Fohlmann [137] vorgeschlagenen Nomenklatur für mögliche Fragmentionen.
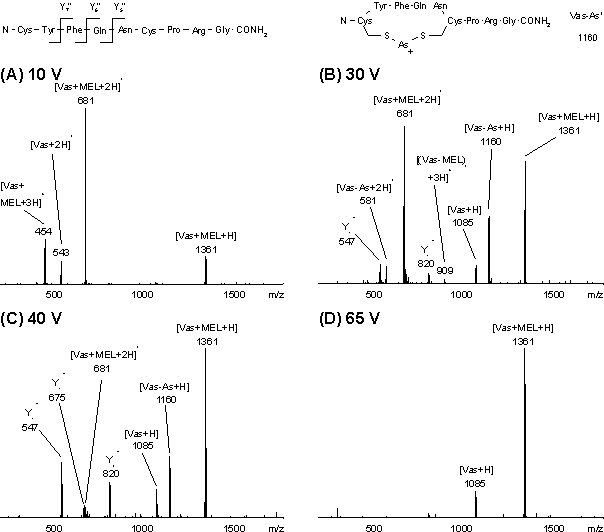
Abb. 54: ESI-Massenspektren von Vas-MEL für verschiedene Repellerspannungen. Die detektierten Fragmente von reduziertem, nicht modifizierten Vasopressin sind in der Primärstruktur gekennzeichnet. Das Fragmention Vas-As+ ist dargestellt.
In Abb. 54 sind die ESI-Massenspektren von Vas-MEL für die Repellerspannungen 10, 30, 40 und 65 V dargestellt. Das Spektrum bei 10 V zeigt das ein-, zwei- und dreifach protonierte Molekülion [Vas+MEL+nH]n+ von Vas-MEL (m/z 454, 681 und 1361), neben geringen Anteilen an zweifach protonierten (m/z 543), nicht modifizierten Vasopressin. Mit zunehmender Repellerspannung verschiebt sich die Ladungsverteilung von Vas-MEL und Vasopressin zu einfach protonierten Molekülionen (m/z 1085 und 1361). Ab 30 V erfolgen Fragmentierungen: Der Bruch der As-C Bindung führt zu dem einfach und zweifach protonierten Fragmention [Vas-As]+ bei m/z 581 und 1160, dessen Struktur inAbb. 54 angegeben ist. Vermutlich erfolgt auch der Bruch der As-S Bindung, da Fragmentionen der Y-Serie des nicht modifizierten Vasopressins bei m/z 820 (Y7),675 (Y6) und 547 (Y5) deutlich zu erkennen sind (Abb. 54, 40 V). Die Fragmentierung führt zum Bruch der Peptidkette zwischen den Cysteinen. Dagegen zeigt Vasopressin mit intakter Disulfidbrücke keine Fragmentierungen der Y-Serie. Daß die Fragmentierung erst nach Abspaltung der MEL-Modifizierung erfolgt, läßt darauf schließen, daß beide Cysteine an der Modifizierung beteiligt sind.
Das Signal bei m/z 908 entspricht dem dreifach protonierten Molekülion von dimeren Vas-MEL. Die Bildung der Dimeren ist abhängig von der Probenpräparation (siehe 2.4.1) und kann durch alle MS-Methoden nachgewiesen werden. Analoge ESI-MS-Analysen wurden für Vas-PAA, Vas-PYR und Vas-A-PAA durchgeführt. In Tab. 5 sind die auftretenden Molekülionen zusammengefaßt.
Zusammenfassend zeigen die Modelluntersuchungen, daß eine nahezu quantitative Umsetzung von vollständig reduziertem Vasopressin durch die Arsonigen Säuren PAA, A-PAA und MEL erfolgt. Aufgrund der langwelligsten und intensivsten UV-Absorption (siehe 2.1.4) und damit der Möglichkeit der UV-spektroskopischen Kontrolle der Modifizierungsreaktion (siehe 2.2.3) wurde ausschließlich MEL für die weiteren Untersuchungen verwendet. Ferner eignen sich alle MS-Methoden (MALDI-, PD-, ESI-MS) grundsätzlich zur Untersuchung von Peptiden und Proteinen, die mit Derivaten der Arsonigen Säure modifiziert wurden. Die ESI-MS-Analyse am Modellsystem Vasopressin liefert durch induzierbare Fragmentierungen zusätzliche Informationen über die Position der Modifizierungsstelle in der Polypeptidkette.
Tab. 5: Experimentell gefundene und berechnete m/z-Werte der Fragment- und Molekülionen der ESI-MS-Analyse von Vas-PAA, Vas-PYR, Vas-MEL und Vas-A-PAA. -M- bezeichnet das modifizierte, -Vas- das nicht modifizierte Vasopressin.
|
Vas-PAAm/z-Wert |
Vas-PYRm/z-Wert |
Vas-MELm/z-Wert |
Vas-A-PAAm/z-Wert |
Zuordnung |
m/z berechnet |
|
- |
- |
454 |
- |
[MH3]3+ |
454 |
|
544 |
544 |
544 |
544 |
[VasH2]2+ |
543 |
|
548 |
- |
549 |
549 |
[Vas-Y5H]+ |
547 |
|
581 |
581 |
581 |
581 |
[Vas-As]2+ |
580 |
|
620 |
621 |
681 |
628 |
[MH2]2+ |
(Abb. 48) |
|
676 |
- |
674 |
675 |
[Vas-Y6H]+ |
674 |
|
- |
698 |
- |
- |
[Vas+2PYR +2H]2+ |
695 |
|
822 |
821 |
820 |
822 |
[Vas-Y7H]+ |
821 |
|
1085 |
1085 |
1085 |
1085 |
[VasH]+ |
1085 |
|
1159 |
1161 |
1159 |
1161 |
[Vas-As]+ |
1159 |
|
1237 |
1238 |
1361 |
1252 |
[MH]+ |
(Abb. 48) |
|
- |
1391 |
- |
- |
[Vas+2PYR +H]+ |
1389 |
|
|
|
|
|
2.4 Untersuchungen zur Stabilität der Reaktionsprodukte von mit Melarsen Oxid modifizierten Modellpeptiden |
|
|
2.2 UV-spektroskopische Untersuchungen zum Reaktionsmechanismus der Modifizierung von Bis-Thiolen mit Melarsen Oxid |
Diese Seite wurde erstellt am 15. März 1998. Letzte Änderungen am 30. Juni 1998 durchPeter Happersberger.