|
|
|
|
|
1.4 Bedeutung, Struktur und biologische Funktion desMakrophagen- Kolonie- Stimulationsfaktors (M-CSF) |
|
|
1.2 Derivate der Arsonigen Säure als chemoselektive Modifizierungsreagenzien |
1.3 Massenspektrometrische Methoden zur Charakterisierung von proteinchemischen Umsetzungen
Die Massenspektrometrie nimmt neben der Röntgenstrukturanalyse und NMR-Spektroskopie eine wichtige Rolle bei der Molekülstrukturaufklärung ein. Die Grundlage der im Vergleich zur NMR-Spektroskopie sehr viel empfindlicheren Methode ist die Bildung von Ionen, die entsprechend dem Verhältnis von Masse zu Ladung (m/z) aufgetrennt und detektiert werden.
Die Anwendung der Massenspektrometrie als Methode zur Strukturanalyse von leichtflüchtigen organischen Molekülen begann in den 60er Jahren mit der Einführung derElektronenstoß-Ionisations-Massenspektrometrie (EI-MS). In der EI-MS werden die Probenmoleküle in der Gasphase durch Elektronenbeschuß (70 eV) ionisiert und nach Beschleunigung durch eine Potentialdifferenz in einem magnetischen Sektorfeld analysiert. Lediglich Moleküle, die eine ausreichende thermische Stabilität und Flüchtigkeit aufweisen können EI-massenspektrometrisch charakterisiert werden. Jedoch können kurzkettige Peptide mit bis zu sechs Aminosäuren nach Derivatisierung [78] ihrer polaren Gruppen per EI-MS analysiert werden. Die dabei auftretenden Fragmentierungen ermöglichen die Bestimmung der Peptidsequenz [79].
Die Entwicklung neuer Ionisations- und Desorptionsmethoden ermöglichte die Charakterisierung von schwerflüchtigen und thermisch instabilen Molekülen. Die im folgenden beschriebenen "schonenden" Ionisationstechniken ermöglichen die massenspektometrische Analyse von Biopolymeren mit Molekulargewichten von mehr als 100 kD. Im Gegensatz zu den Methoden der Gasphasenionisation erfolgt die Ionisation der Probenmoleküle in kondensierter Phase.
Mit der Einführung der Fast-Atom-Bombardment-Massenspektrometrie(FAB-MS) anfang der 80er Jahre war die Analytik von Peptiden ohne vorherige Derivatisierung möglich [80]. Der Ionisations- und Desorptionsprozeß ist schematisch in Abb. 11 dargestellt. Die Probenmoleküle sind in einer viskosen Lösungsmittelmatrix (z.B. Glycerin, 3-Nitrobenzylalkohol) gelöst. Die Desorption der in der Matrix bereits ionisierten Probenmoleküle erfolgt durch Beschuß mit Neutralatomen oder Primärionen (z.B. Ar, Xe, Cs+). Diese dringen in die Matrix ein und verursachen die Emission von Sekundärionen. Trotz der hohen Energien der Primärteilchen (10-20 keV) läuft die Desorption dieser Sekundärionen sehr schonend ab, da durch die in der Matrix ablaufende Stoßkaskaden nur ein sehr schwacher Energietransfer stattfindet [81]. Neben den desorbierten Molekülionen treten Matrixclusterionen auf. Nach Beschleunigung durch eine Potentialdifferenz werden die Sekundärionen in einem magnetischen und/oder elektrischen Sektorfeldgerät aufgetrennt und detektiert. Die üblicherweise verwendeten Geräte erlauben eine Detektion von Ionen bis zu m/z-Wert von 6000. Bei einer Auflösung (m/m > 10000) ist damit die Bestimmung der Isotopenverteilung möglich.
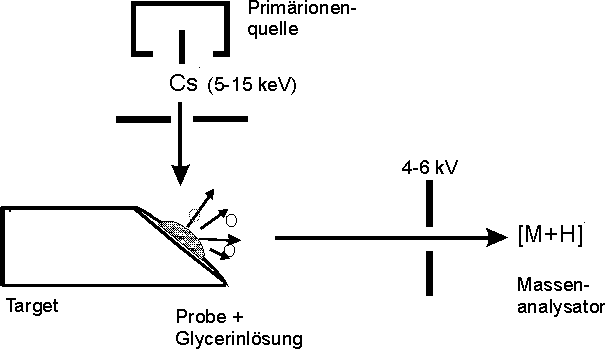
Abb. 11: Schematische Darstellung der Ionenquelle eines FAB-Massenspektrometers. Die Desorption der Probenmoleküle erfolgt durch Beschuß des Matrix-Proben-Gemischs mit energiereichen Primärionen (Cs+).
Bei der Plasmadesorptionsmassenspektrometrie (PD-MS) [82, 83] erfolgt die Desorption und Ionisierung der Probenmoleküle durch Beschuß der Probe mit hochernergetischen Tochternukliden von252Californium.252Cf zerfällt mit einer Halbwertszeit von 2.65 Jahren zu 97% durch -Zerfall und zu 3% unter der diametralen Aussendung von hochgeladenen Nukliden mit Massen zwischen 100 und 150 amu (z.B.142Ba18+ und106Tc22+). Diese hochenergetischen Nuklide (80 bis 120 MeV) dienen zum einen als Signalgeber für den Startdetektor und zum anderen zur Desorption und Ionisation der Probenmoleküle (siehe Abb. 12) Die Probe ist auf einer nitrocellulosebeschichteten, aluminiumbedampften Terephthalatfolie adsorbiert. Das Nuklid bewirkt beim Durchschuß dieser Folie ein Plasma mit einer Temperatur von 10000 K und erreicht damit eine schnelle Desorption von neutralen und geladenen Molekülen bis zu 50 kD [81, 83]. Die Ionisierung erfolgt äußerst schonend und ohne wesentliche Fragmentierung der Probenmoleküle . Die desorbierten Sekundärionen werden in einem elektrischen Feld (10-20 kV) beschleunigt und gelangen nach Durchlaufen einer feldfreien Strecke zum Stopdetektor. Die Zeitdifferenz zwischen dem Start- und Stopsignal wird über die lineare Beziehung der Quadratwurzel des m/z-Werts und der Flugzeit in ein Massenspektrum umgewandelt. Der Flugzeitanalysator (TOF, time of flight) hat den Vorteil eines prinzipiell nicht limitierten Massenbereichs, kann jedoch wegen seiner eingeschränkten Massenauflösung (m/m 500) keine Isotopenverteilung wiedergeben. Da nur ein sehr kleiner Teil der auf dem Probenträger absorbierten Substanz durch die Messung verbraucht wird, können auf dem Probenträger weitere chemische und enzymatische Reaktionen durchgeführt und die Probe anschließend vermessen werden.
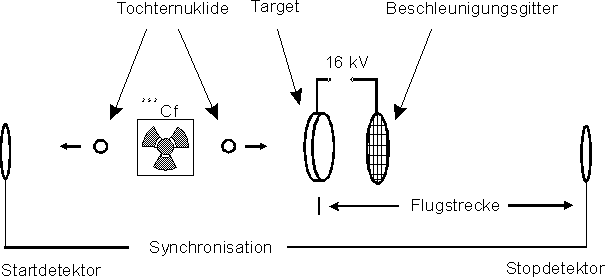
Abb. 12: Schematische Darstellung eines PD-Massenspektrometers: Die Nuklide des Zerfalls von252Cf (z.B.142Ba18+ und106Tc22+) erzeugen synchron das Startsignal und die Ionisation/Desorption der Probenmoleküle.
Mit der Elektrospray-Ionisations-Massenspektrometrie (ESI-MS) gelang der Durchbruch zur direkten massenspektrometrischen Analyse von hochmolekularen Proteinen [84, 85]. Die Proteinlösung wird hierbei unter Normaldruck durch eine Kapillare kontinuierlich in die Ionenquelle eingeleitet (sieheAbb. 13). An der Kapillarspitze wird ein hohes elektrisches Potential (2-3 kV) angelegt, so daß ein Aerosol aus elektrische geladenen Tröpfchen entsteht. Durch die Desolvatisierung im Vakuum steigt die Oberflächenfeldstärke so weit an, daß die Oberflächenspannung überwunden wird und die Tröpfchen zerplatzen. Dieser Prozeß wiederholt sich für die neu entstandenen kleineren Tröpfchen, so daß mehrfach geladene Molekülionen in die Gasphase freigesetzt werden.
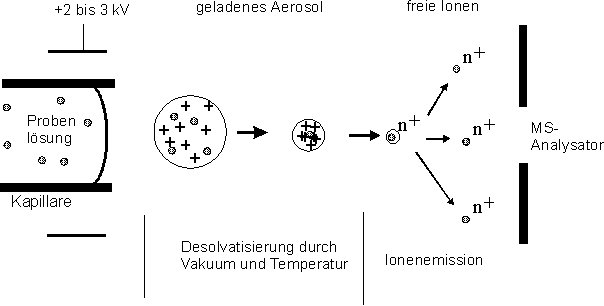
Abb. 13: Schematische Darstellung der Ionenbildung der ESI-Massenspektrometrie: Die gelöste Probe bildet durch ein angelegtes Potential geladene Tröpfchen, die durch Desolvatisierung im Vakuum zur Emission von mehrfach geladenen Probenmolekülen führt.
Die Massenanalyse dieser Molekülionen erfolgt meist mit einem Quadrupolanalysator. Charakteristisch für die ESI-MS ist das Auftreten von kontinuierlichen Serien mehrfach geladener Molekülionen, die die genaue Massenbestimmung von Molekülen mit hohen Molekulargewichten ermöglicht. Die Ionisierung bei der ESI-MS verläuft äußerst schonend, so daß eine Charakterisierung nicht-kovalenter Komplexe möglich ist [86-88]. Weiterhin erlaubt die ESI-MS, zwischen nativen und denaturierten Proteinstrukturen aufgrund ihrer unterschiedlichen Ladungsstruktur zu unterscheiden [89].
Mit der seit 1988 eingeführtenMatrixunterstützten-Laserdesorptions-Massenspektrometrie(matrix-assisted-laser-desorption-ionization (MALDI)-MS) steht eine weitere massenspektrometrische Methode zur Molekulargewichtsbestimmung von Proteinen mit mehr als 100 kD zur Verfügung [90] (sieheAbb. 14). Die wäßrige Probe wird dazu in einem hohen molaren Überschuß mit einer im ultravioletten Spektralbereich absorbierenden Substanz (Matrix) cokristallisiert. In der Praxis gängige Matrizes sind Zimtsäurederivate, wie z.B. -Cyano-4-hydroxyzimtsäure (HCCA). Durch Beschuß dieser Matrix-Proben-Mikrokristallite im Hochvakuum mit einem Laser (z.B. 337 nm, Stickstofflaser) erfolgt die Desorption und Ionisation der Probenmoleküle. Die Art der Matrix spielt bei der MALDI-Massenspektrometrie eine entscheidende Rolle, da sie wahrscheinlich als Energieüberträger, Protonenpool und zur Isolierung der Probenmoleküle dient, sowie die Pyrolyse der Probe verhindert [91]. Der Mechanismus der Desorption und Ionisation ist noch Gegenstand der Forschung. Die so erhaltenen Ionen werden mit Hilfe einer Potentialdifferenz von 10-30 kV beschleunigt und mittels Flugzeitanalysator massenspektrometrisch charakterisiert. Die Kalibrierung des MALDI-Massenspektrometers erfolgt mit einem Protein, dessen Molekulargewicht bekannt ist. Die Genauigkeit der Massenbestimmung beträgt etwa 0.01% bis 0.1% [92].
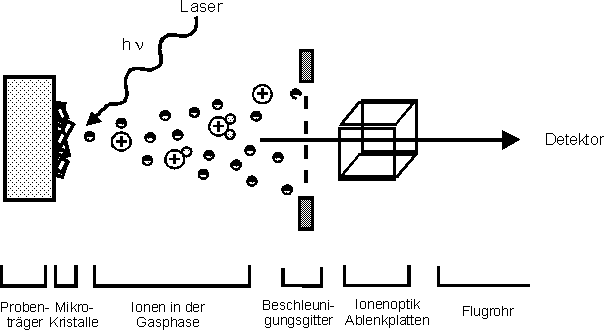
Abb. 14: Schematische Darstellung des MALDI-Massenspektometers: Durch den Laserbeschuß des Matrix-Proben-Gemischs erfolgt sowohl der Start der Messung der Flugzeit, als auch die Desorption/Ionisation der Probenmoleküle. Die desorbierten und ionisierten Moleküle werden im elektrischen Feld beschleunigt und die m/z-Werte im Flugzeitanalysator bestimmt.
Die MALDI-MS-Methode zeigt für die Analyse von Peptidgemischen ausgezeichnete Empfindlichkeiten und nur geringe Unterdrückungseffekte. Ein weiterer Vorteil für die Proteinanalytik ist die geringe benötigte Probenmenge (1-20 pmol) [93] und die schnelle praktische Durchführung der Messung. Die geringe Auflösung des TOF-Analysators (m/m 500), sowie unspezifische Adduktbildung u.a. mit der Matrix ermöglichen nur die Bestimmung des durchschnittlichen Molekulargewichts des Analyts.
|
|
|
|
|
1.4 Bedeutung, Struktur und biologische Funktion desMakrophagen- Kolonie- Stimulationsfaktors (M-CSF) |
|
|
1.2 Derivate der Arsonigen Säure als chemoselektive Modifizierungsreagenzien |
Diese Seite wurde erstellt am 15. März 1998. Letzte Änderungen am 30. Juni 1998 durchPeter Happersberger.