|
|
|
|
|
1.2 Derivate der Arsonigen Säure als chemoselektive Modifizierungsreagenzien |
1. Einleitung und Problemstellung
1.1 Cysteinylthiolgruppen und Disulfidstrukturen in Proteinen
1.1.1 Bedeutung der Cysteinylthiolgruppen für Proteinstruktur und -funktion
Neben den Nukleinsäuren stellen die Proteine die wichtigste Gruppe an Biomakromolekülen dar. Fast alle biochemischen Prozesse in Lebewesen werden durch Proteine reguliert. So katalysieren Enzyme chemische Reaktionen, die unter physiologischen Bedingungen sonst nicht ablaufen würden. Des weiteren dienen Proteine als Speicher- und Transportsysteme z.B. von Elektronen im Photosyntheseprozeß oder von kleinen organischen Molekülen wie z.B. der Häm-Gruppe im Hämoglobin. Proteinhormone wie die Cytokine vermitteln den Informationsaustausch zwischen Zellen des Immunsystems. Auch die Expression bestimmter Gene wird durch DNA-bindende Proteine kontrolliert. Die Vielzahl der Proteinfunktionen beruht v.a. auf der Ausbildung einer unterschiedlichen dreidimensionalen Struktur (Tertiärstruktur) der einzelnen Makromoleküle, dabei spielen die unterschiedlichen chemischen Eigenschaften der Aminosäuren eine nur untergeordnete Rolle [1, 2]. Die Tertiärstruktur aller Proteine wird in erster Linie durch die Primärstruktur festgelegt. Die vollständige Primärstruktureines Proteins wird durch die Aminosäuresequenz und die posttranslationalen Strukturmodifikationen bestimmt. Zu den letzteren zählen die Glykosylierung oder Phosphorylierung bestimmter Aminosäuren, sowie die Ausbildung (kovalenter) Disulfidstrukturen. Die räumliche Anordnung linear aufeinanderfolgender Aminosäurereste, die eine gleiche Konformation besitzen, wird durchSekundärstrukturelemente beschrieben. Man unterscheidet dabeihelicale Bereiche von planaren Faltblattstrukturen, die untereinander durch Loops verbunden sein können [1, 2]. Selbst bei Kenntnis der vollständigen Primärstruktur, ist es derzeit nicht möglich, das exakte Faltungsmuster der Sekundärstrukturelemente und die räumliche Plazierung der Aminosäurenseitenketten, also der Tertiärstruktur, vorherzusagen.
Die Aminosäure Cystein ist wesentlich an der Ausbildung der Proteinfunktion und -struktur beteiligt. Man kann vier Bereiche unterscheiden, in denen Cysteinseitenketten strukturell und funktionell von Bedeutung sind. Als Aminosäure mit der reaktivsten Seitenkette (pKa der Thiolgruppe 9.0-9.5 [1]) finden sich Cysteinylthiolgruppen im aktiven Zentrum vieler Enzyme. Eine erste Proteinklasse, in denen Cysteinthiolgruppen von enzymatischer Bedeutung sind, sind die sogenannten Thiolproteasen. Diese gehören wie die Metallo-, Serin- und Carboxypeptidasen zur Gruppe hydrolytischer (proteolytischer) Enzyme [3]. Zweitens besitzen Thiol-Oxidoreduktasen, wie z.B. das Thioredoxin, im aktiven Zentrum zwei Cysteinylreste. Der Wechsel zwischen oxidierter Disulfidstruktur und reduzierter Dithiolform bewirkt einen Transport von Reduktionsäquivalenten in Form von Wasserstoffatomen [4]. Ferner fungieren Cysteinylthiolgruppen als Träger posttranslationaler Strukturmodifikationen. So sind die zwei vicinalen Cysteinreste im Lungensufactant-Protein SP-C fettsäureacyliert [5] Eine weitere posttranslationale Strukturmodifikation ist die Ausbildung von Disulfidbrücken (Cystinylgruppen) und geschieht in eukaryontischen Zellen als einer der letzten Schritte der Proteinbiosynthese. Durch die Ausbildung der Disulfidbrücken kann die Tertiärstruktur des Proteins und damit seine Funktion bestimmt werden. Wie De- und Renaturierungsexperimente an der Ribonuklease A zeigten, ist die Tertiärstruktur eines Proteins zwar in der Aminosäuresequenz enthalten, jedoch erfolgt erst durch die korrekte Schließung der Disulfidbrücken eine kovalente Fixierung der Proteinstruktur [6].
1.1.2 Bedeutung der Disulfidstrukturen in der Proteinfaltung
Die Ausbildung von Disulfidstrukturen in Proteinen erfolgt erst in späteren Stadien der in vitro Faltungsreaktion von denaturierten Proteinen. Aufgrund der astronomisch großen Zahl an möglichen Konformationen einer entfalteten Polypeptidkette kann die Rückfaltung zum nativen Protein nicht durch zufälliges "Ausprobieren" aller Konformationen erfolgen [7]. Vielmehr nimmt man für die möglichen Faltungsabläufe (folding pathways) hierarchische Abfolgen von Konformationsänderungen an. Derzeit werden verschiedene Modelle diskutiert [8-10]. Im Modell des"hydrophobic collapse" nimmt man an, daß sich die Polypeptidkette des ungefalteten (denaturierten) Protein sehr schnell bis annähernd zur endgültigen Dichte des Proteins verknäult, da so die hydrophoben Aminosäuren schnell untereinander Wechselwirkungen eingehen können. Die treibende Kraft der Proteinfaltung sind die hydrophoben Wechselwirkungen der hydrophoben Aminosäuren der Polypeptidkette mit dem Lösungsmittel. Ausgehend von diesem Zustand würde dann die Umorganisation zur korrekten Sekundär- und Tertiärstruktur erfolgen [11]. Dagegen geht man im "framework model"davon aus, daß die Faltung des denaturierten Peptids mit der zufälligen Bildung kurzer Abschnitte von Sekundärstrukturen beginnt [12]. Diese Sekundärstrukturen können als Nuclei (Gerüst) weitere Regionen innerhalb der Polypeptidkette stabilisieren. Mehrere Nuclei mit beinahe nativer Struktur wachsen wahrscheinlich durch Diffusion, Zufallskollision und Adhäsion. Die Stabilitäten dieser geordneten Regionen nehmen mit ihrer Größe zu, so daß in kooperativer Weise (fast) native Domänen des Proteins gebildet werden [13]. In Multidomänen-Proteinen lagern sich die Domänen zu locker organisierten, aber fast nativen Untereinheiten ("molten globules") zusammen, deren hydrophoben Seitenketten noch dem Lösungsmittel ausgesetzt sind [14,15]. Durch relative kleine Konformationsänderungen der Polypeptidkette wird die endgültige native Konformation erreicht [2].
Die geschwindigkeitsbestimmenden Schritte der Rückfaltungsreaktion vieler Proteine sind die Isomerisierung der Prolinpeptidbindung [16] und die Ausbildung der Disulfidstrukturen [17]. Für die oxidative in vitroRückfaltung werden deshalb weitere, meist niedermolekulare Thiolkomponenten, wie das Tripeptid Glutathion (L-gamma-Glutamyl-L-cysteinyl-glycin, GSH) in oxidierter (Bis-(L-gamma-Glutamyl-L-cystinyl-bisglycin), GSSG) und reduzierter Form (GSH) zugesetzt, um die Thiol-Disulfidgleichgewichtseinstellung zu beschleunigen [18]. Die Ausbildung der Disulfidbindungen im Protein vollzieht sich unter der Gleichgewichtseinstellung einer Vielzahl an Redoxreaktion zwischen Proteinthiolen und der zusätzlichen Thiolkomponente (siehe Abb. 1 A). Neben demintermolekularen Thiol-Disulfidaustausch spielen nochintramolekulare Austauschreaktionen zwischen bereits gebildeten Disulfidbindungen und freien Thiolgruppen innerhalb des Proteins eine Rolle (siehe Abb. 1 B). Beide Reaktionen korrigieren "falsch" ausgebildete Disulfidstrukturen und führen letztendlich zum nativen Protein [10,19].
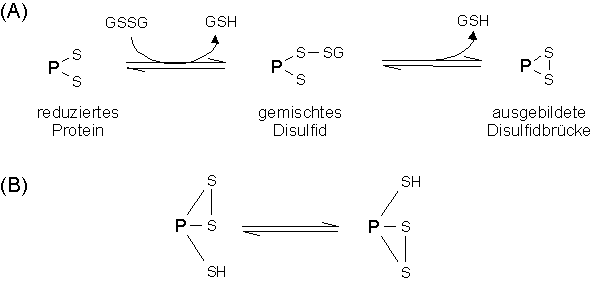
Abb. 1: (A) Redoxgleichgewichte zwischen Proteinthiolen und der Thiolkomponente Glutathion in reduzierter (GSH) und oxidierter Form (GSSG). (B) Intramolekulare Austauschreaktionen zwischen bereits gebildeten Disulfidbindungen und freien Thiolgruppen innerhalb des Proteins (P).
In in vivo Faltungsreaktionen katalysierenProtein-Disulfid-Isomerasen die Disulfidaustauschreaktion [20]. Weitere an in vivo Proteinfaltungen beteiligte Proteine sind die Peptidyl-Prolyl-cis/trans-Isomerasen[21] und die als Chaperone bezeichneten Proteine, welche sich während der Proteinbiosynthese an die hydrophoben Sequenzbereiche der noch "unvollständigen" Aminosäurekette anlagern und so eine Aggregation und Mißfaltung zu nicht nativen Strukturen verhindern [22, 23]. Gerade diese Nebenreaktionen (Aggregation, Mißfaltung) der Proteinfaltung können bei der in vitro-Rückfaltung von gentechnisch hergestellten eukaryontischen Proteinen zu hohen Ausbeuteverlusten führen [24, 25]. Des weiteren wird vermutet, daß sich eine Vielzahl von Krankheiten auf der Mißfaltung (z.B. Cystische Fibrose) oder Aggregation von Proteinen (z.B. Scrapie, Creutzfeld-Jakob oder Alzheimer) begründen [26].
Ausgehend von diesen Faltungsmodellen ermöglicht die Charakterisierung der in vitro-teilgefalteten Intermediate Rückschlüsse auf den Mechanismus der Proteinrückfaltungsreaktion. Wesentliche Ansätze zur Charakterisierung von Faltungsintermediaten sind Mutageneseexperimente [27, 28], Untersuchungen an Peptidmodellen [29] und H/D-Austauschexperimente mit 1H-NMR-Analysen [30] und/oder MS-Analysen [31]. Ein wesentliches Problem der Analyse der Rückfaltungsreaktion ist die Stabilisierung der instabilen Faltungsintermediate. Aufgrund des chemischen Charakters der Cysteinylthiolgruppe ist es für Proteine mit Disulfidbrücken möglich, den zeitlichen Verlauf des Thiol-Disulfidaustausches zwischen dem zusätzlich eingesetzten Thiol und dem Protein zu beobachten. Durch "Quechnen" des Thiol-Disulfidaustausches durch Alkylierung (s.u.) der Cysteinylgruppen oder Ansäuern der Reaktionslösung können die so erhaltenen Faltungsintermediate stabilisiert, und damit isoliert und charakterisiert werden [32, 33]. Dabei nimmt man an, daß jede unterscheidbare disulfid-verbrückte Spezies eine Teilgruppe der Konformationen repräsentiert, die die rückzufaltende Polypeptidkette einnehmen kann. Durch das Verfolgen des zeitlichen Verlaufs dieser verschiedenen Spezies kann der mutmaßliche Konformationsablauf des renaturierenden Proteins abgeleitet werden [1]. In neueren Untersuchungen wurden so stabilisierte Faltungsintermediate von Myoglobin [34], Insulin [35], RNAse A [36] und rhM-CSF (siehe 1.4.3) [37] mit massenspektrometrischen Methoden untersucht.
1.1.3 Proteinchemische Methoden zur Charakterisierung von Cysteinylthiolgruppen
Zur Alkylierung und damit Stabilisierung der cysteinylhaltigen Faltungsintermediate stehen eine Reihe von proteinchemischen Umsetzungen zur Verfügung. Da die Sulfhydrylgruppe die nukleophilste Seitenkettenfunktion in Proteinen darstellt, basieren die meisten bisher zur Modifikation von Cysteinylresten verwendeten Verfahren auf einem nukleophilen Angriff des Schwefels auf ein positiviertes oder ungesättigtes Kohlenstoffatom. Die Alkylierung der Thiolgruppe durch Iodessigsäure oder Iodacetamid (IAA) [38,39] erfolgt über eine Sn2-Substitution am Halogen-Kohlenstoff durch die Cysteinylthiolgruppe. Dabei entstehen S-Carboxymethyl- bzw. S-Carboxamidomethyl-Derivate. Die Umsetzung mit 4-Vinylpyridin (VP) stellt analog der Michael-Addition eine nukleophile Addition des Cysteinylthiols an die durch den Pyridinring aktivierte Vinylseitenkette dar und führt zu S-pyridylethylierten Reaktionsprodukten [39,40]. Ebenfalls durch nukleophile Addition an die Doppelbindung erfolgt die Alkylierung der Cysteinylthiolgruppe durch N-Ethylmaleinimid [41]. Diesen Alkylierungsmitteln gemeinsam ist die Durchführung der Modifizierungsreaktion in wäßriger, schwach alkalischer Lösung und die Anfälligkeit für Nebenreaktionen mit anderen nukleophilen Seitenketten. So beobachtet man zusätzliche Alkylierungen des N-Terminus und der Lysin--Aminogruppe [42]. Durch massenspektrometrische Molekulargewichtsbestimmung (siehe 1.3) der so modifizierten Faltungsintermediate ist der Umsetzungsgrad der proteinchemischen Modifikation und damit die Anzahl an noch nicht ausgebildeten Disulfidbrücken des Proteins bestimmbar. Insbesondere ermöglicht die Kombination von proteinchemischen Umsetzungen, proteolytischen Abbaureaktionen an Protein-Faltungsintermediaten und die massenspektrometrische Charakterisierung der Peptidfragmente die Identifizierung der modifizierten Cysteinreste. Somit kann die Reihenfolge der ausgebildeten Disulfidbindungen während der Rückfaltungsreaktion ermittelt werden [37].
Ein weiteres Modifizierungsreagenz ist die Verbindung 2,2'-Dinitro-5,5'-dithiodibenzoesäure (DTNB, Ellmans Reagenz). Das symmetrische Disulfid DTNB wird durch nukleophilen Angriff einer Cysteinylthiolgruppe zum asymmetrischen Cysteinyl-S-S'-5-(2-nitrobenzoat) umgesetzt. Das durch den Disulfidaustausch entstandene 2-Nitro-5-thiobenzoat-Anion wird UV-spektroskopisch quantitativ bestimmt. Die Modifizierungsreaktion eignet sich zur Bestimmung der Thiolkonzentration. [43]. Jedoch kann das 2-Nitro-5-thiobenzoat-Anion mit Disulfidbrücken im Protein zu gemischten Disulfiden reagieren, wodurch eine Differenzierung zwischen Cysteinyl- und Cystinylresten erschwert wird.
In neueren Untersuchungen in unserer Arbeitsgruppe wurde Phenylarsenoxid (PAA; auch PAO, zur Nomenklatur siehe 1.2.1) alstertiärstrukturselektives Modifizierungsreagenz eingesetzt. Die chemisch selektive Modifizierung von Biscysteinylpeptiden mit PAA über einen weiten pH-Bereich und dieerhöhte Stabilität makrozyklischer Arsonigsäuredithioester (siehe 1.2.3) gegenüber offenkettigen konnte massenspektrometrisch nachgewiesen werden. Massenspektrometrische Peptide-Mapping-Analysen von mit PAA modifizierten Thioredoxin f bestätigten die tertiärstrukturselektive Modifizierung der im aktiven Zentrum liegenden Cysteine 49 und 52, während das Cystein 76 nicht modifiziert gefunden wurde [44,45].
Tab. 1: Bisher angewendete proteinchemische Umsetzungen zur Modifikation von Cysteinylthiolgruppen in Proteinen.
|
Modifizierungsreagenz |
pH-Wert während der Umsetzung/ Selektivität |
bekannte Nebenreaktionen |
|
Iodessigsäure / Iodacetamid |
pH > 7 / Cysteinylthiole |
N-Terminus, Lysin--NH2 |
|
4-Vinylpyridin |
pH > 7 / Cysteinylthiole |
N-Terminus, Lysin--NH2 |
|
N-Ethylmaleinimid |
pH > 7 / Cysteinylthiole |
N-Terminus, Lysin--NH2 |
|
2,2'-Dinitro-5,5'-dithiodibenzoesäure |
pH > 7 / Cysteinylthiole |
Reaktion mit Cystinylgruppen |
|
Phenylarsenoxid |
pH 2-8 /Biscysteinylthiole |
- |
Aufgrund der hier beschriebenen Eigenschaften eignen sich Arsenverbindungen prinzipiell zur Charakterisierung von Proteinthiolgruppen. Insbesondere scheinen Phenylarsenoxid-Derivate in diesem Zusammenhang interessant, da sie verbrückende Strukturen mit zwei Cysteinylfunktionen in Proteinen bilden können. Dadurch wäre eine Stabilisierung von Faltungintermediaten möglich, in denen zwei Cysteine noch keine Disulfidbrücke gebildet haben, sich aber räumlich so nahe stehen, daß sie durch die Arsenderivate verbrückt werden können. Umgekehrt würde keine Arsenverbrückung für räumlich weit voneinander entfernte Cysteine erfolgen. Im Vergleich zu den nicht tertiärstrukturselektivenMonocysteinyl-Alkylierungsmittel (IAA, VP) wäre eine weitere Unterscheidung der Ausbildungsreihenfolge der Disulfidstrukturen während der Faltung von Proteinen denkbar.
|
|
|
|
|
1.2 Derivate der Arsonigen Säure als chemoselektive Modifizierungsreagenzien |
Diese Seite wurde erstellt am 15. März 1998. Letzte Änderungen am 30. Juni 1998 durchPeter Happersberger.